Dr. Clare Craig from the HART group explains the clinical trial used to justify vaccinating kids
Steve Kirsch | June 19, 2022
The HART group is a group of highly respected independent doctors and scientists. My friend, Professor Norman Fenton, is a member of this group.
In this 4 minute video, Dr. Clare Craig, co-chair of the HART group, explains the clinical trial that was used to justify vaccinating our kids. She was appalled.
The only conclusion you can draw after watching this video is that the people running the FDA, CDC and the members of the outside committees approving these vaccines are either completely incompetent or totally bought off.
Everyone should watch this video. It should be required viewing for any parent who is considering vaccinating their child.
Here is the report Pfizer submitted to the FDA referenced in her video. You can see the numbers on page 39 (look in the column headings for the N= numbers).
Developmental Disorders in Babies born to Vaccinated Mothers?
Pfizer wants Babies to be Exposed to SIX Vaccine Shots!
By Igor Chudov | June 10, 2022
I will explain that
- Children of Covid vaccinated mothers were never tested for developmental disorders
- CDC recently revised and lowered developmental milestones, and removed some entirely
- Newly born babies will be exposed to SIX doses of mRNA vaccines if the FDA’ approves the Pfizer vaccine.
An interesting article came out:

This article found that at one year of age, babies born to mothers who had COVID (not vaccine), had a roughly twice-higher rate of neurodevelopmental disorders:
those born to the 222 mothers with a positive SARS-CoV-2 polymerase chain reaction test during pregnancy were more likely to receive a neurodevelopmental diagnosis in the first 12 months after delivery, even after accounting for preterm delivery.
Considering that COVID is a bad disease for a sizable minority of people, there is no surprise. Covid is bad and gives people all sorts of problems. Then I started thinking: a lot of adverse effects of Covid vaccines mimic the adverse effects of Covid. The younger is the vaccine recipient, the worse some effects of vaccination (such as myocarditis) are.
A great number of expectant mothers received up to three Covid vaccine shots during pregnancy. Did anyone bother testing one-year-old children of vaccinated and boosted (during pregnancy) women for neurodevelopmental disorders, before approving this vaccine for all pregnant women?
The question is, obviously, rhetorical, since “mRNA Babies” of triple-vaxed-during-pregnancy mothers are only beginning to get born right now and are at most a few months old. Not one such baby reached a year of age. So nobody tested them for developmental disorders at one year of age, before approving the three vaccine shots for expectant mothers.
The usual argument of vaccinators that “since Covid does it too, you should take the vaccine” does not hold water. To a woman who decided to take the vaccine, the probability of getting a vaccine is 100%. The probability of her getting Covid is much less. In the above study, out of 7,772 births, only 222 (2.8%) were exposed to Covid during pregnancy. So while vaccination is 100% guaranteed for those who elect to vaccinate, the chance of Covid is over 30 times less likely. And the “vaccine” does not prevent Covid anyway and does not even reduce the viral load.
There is literally zero data on one-year-old children of triple-vaccinated mothers because the oldest ones are 3-4 months old as of today.
However, there are disturbing developments regarding newborns. Vaccination does seem to have an effect on births and pregnancies.
The best data I found regarding recently born newborns happens to come from Scotland. They have an interesting “wider impacts” page that I am quoting below.
Infant deaths are way above average and exceeded “Alert Limits” twice.
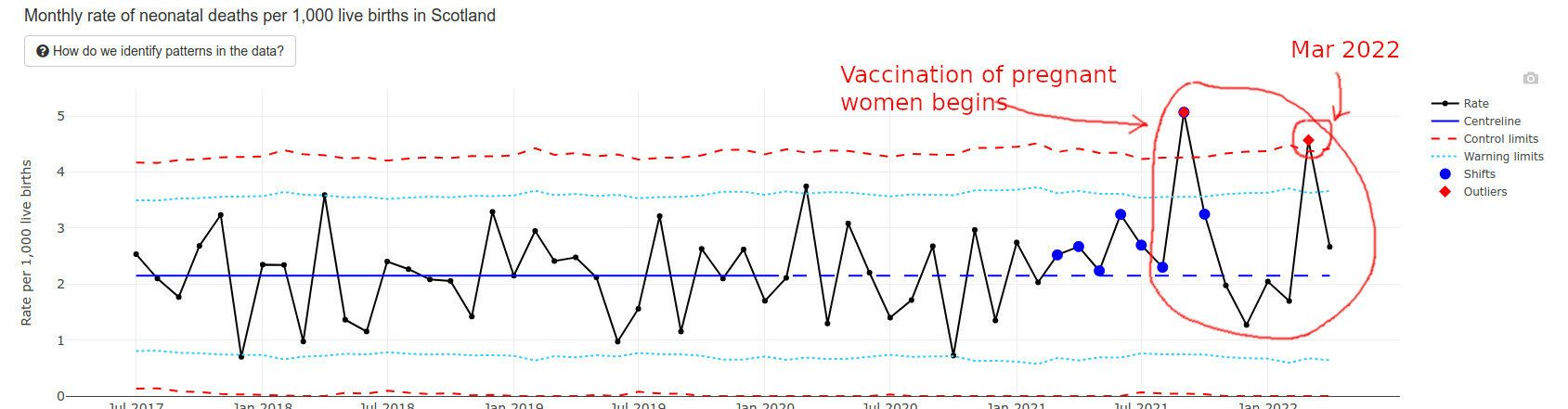
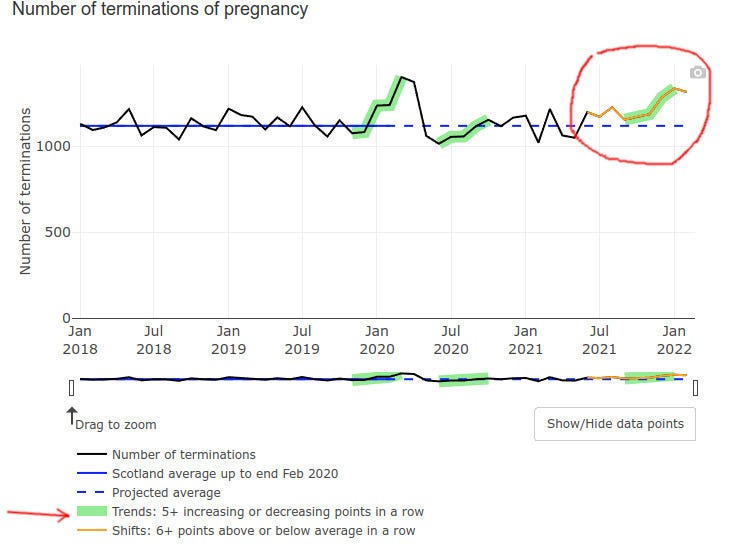
Even pregnancy terminations went up at the end of 2021, possibly but not certainly explained by prenatal vax problems:
Low Apgar score births (for those readers who do not have kids, Apgar score is how healthy is the baby at birth, the best being a score of 10) triggered a green alarm signal:
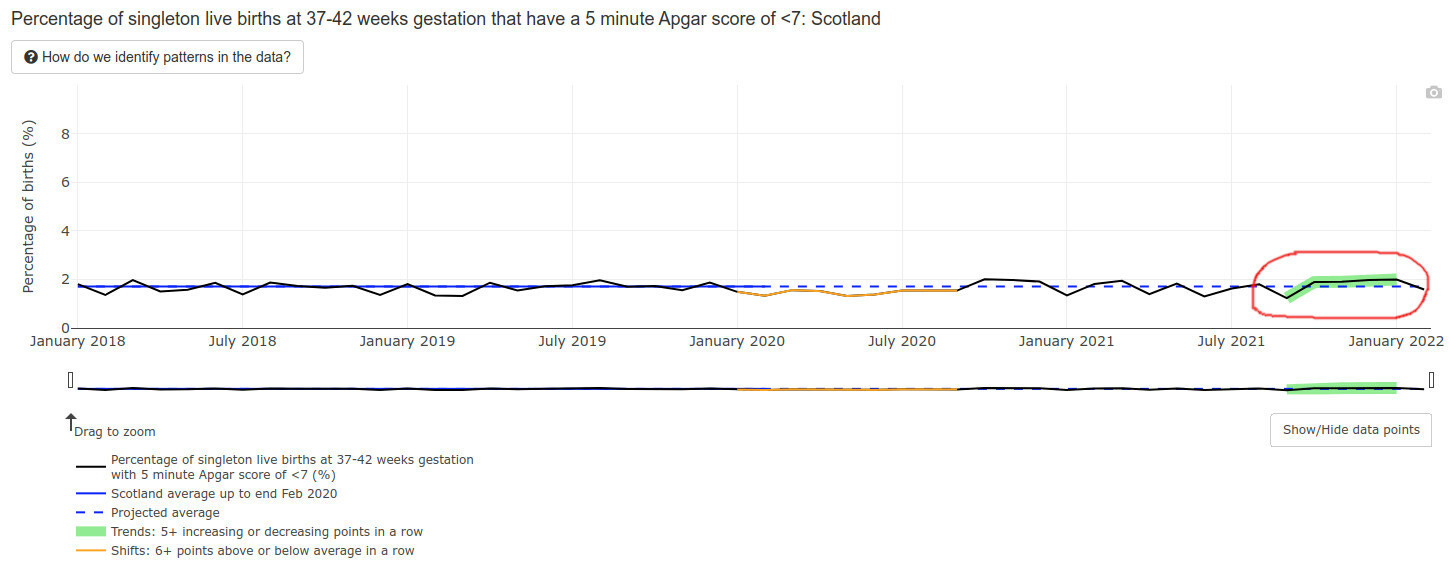
Mind you, an Apgar score is also a developmental evaluation of sorts — at 5 minutes after birth. What will happen to the developmental milestones of those lucky babies of vaccinated mothers, who survived the pregnancies, did not die postnatally, and lived to one year of age? I literally have no idea and nobody else in the world does — the time has not passed yet.
The data we have is NOT encouraging.
CDC Solution: Remove and Lower Milestones
The CDC possibly caught a whiff of this, because in February of 2022 they literally removed half the developmental milestones, bumped some others to higher ages, and lowered standards for yet more of them. (Hat tip @CLesterwood)
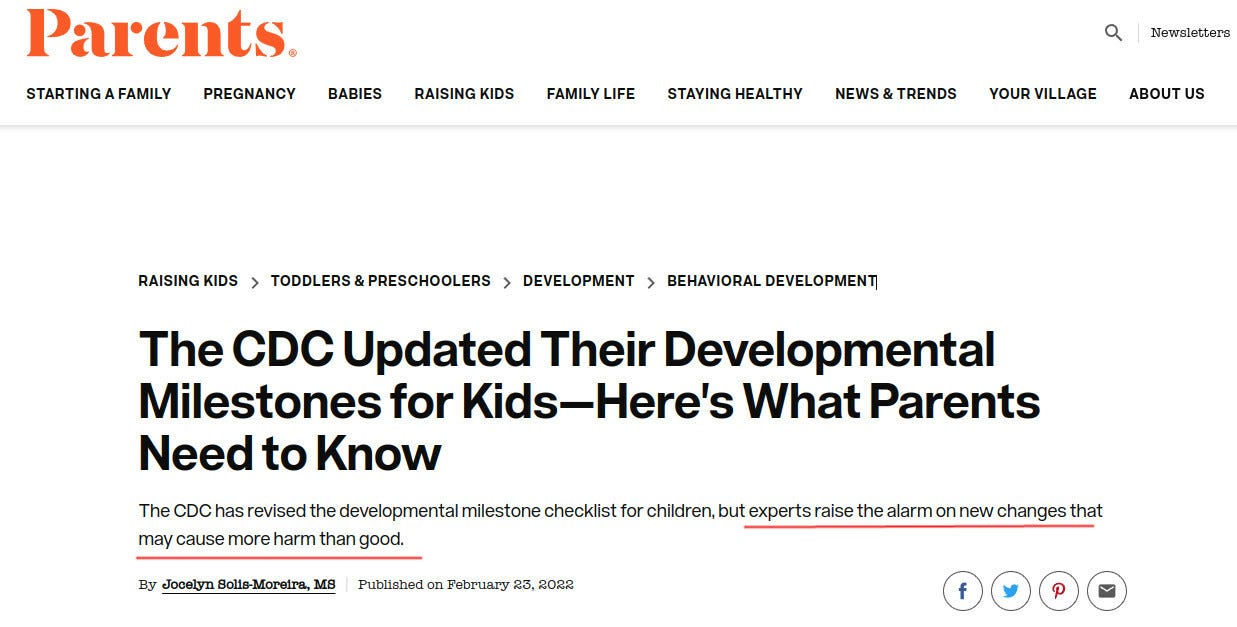
About one-third of milestones like fine motor skills have been bumped up to older ages. Because of the setback, children may worsen their developmental delay, making it harder to provide early intervention, explains Jessica Hatfield, MS, OTR/L, a pediatric occupational therapist for TheraTree Pediatric Therapy.

Removing crawling as a milestone??? Are you kidding us? For those of my readers who are parents, do you think that crawling is unimportant as a milestone?
Vaccinated Infants Exposed to SIX Doses of Covid Vaxx in a Year!
Imagine a vaccine enthusiast mother, who gets three doses during her pregnancy. Say, two doses during month 4 and one during the last week of pregnancy. The unborn baby is, obviously, exposed to all that.
Then the baby is born.
If the June 14-15 FDA meeting goes as planned, FDA will approve a three-dose Pfizer vaccine for infants and toddlers. So shortly after being exposed to THREE doses of mRNA vaccines prenatally, the recently born 6 months old baby will get THREE MORE Pfizer mRNA shots.
That’s a total of, drumroll, six spike protein, and nanoparticle exposures. For a tiny newborn, all during one first year of her life.
And what if the mom has several Covids while being pregnant and vaccinated?
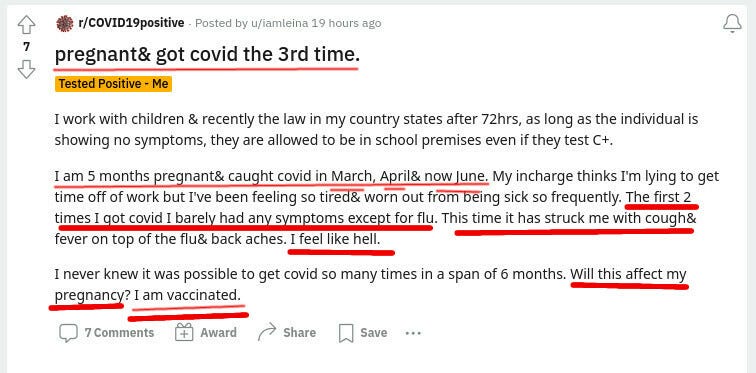
They will ask the mom to vaccinate the baby regardless of those covid infections. This literally amounts to six doses within a year or close to, without even counting actual covids that the vaxed moms have. Pfizer will make $132 from these six shots. Not sure if the baby will eventually need much more expensive treatments.
Do you think that it is a little bit too much? Do you think Pfizer or the FDA care?
Injecting 6 mo. olds to 5yo’s? – NO!
Murder has no statute of limitations
By Coquin de Chien | June 13, 2022
The United States Government, at the behest of Pharma oligarchs and government employees who own stock in the Pharma companies, hopes to approve an amendment to the EUA (Emergency Use Authorization) to inject babies 6-months-old to toddlers 4-years-old with the C19 faux-vaccine.
Before the committee meets to recommend the amendment, the FDA allows people to comment on the FDA government web site. One such comment was provided to this author and is offered to you below. The United States of America is indeed facing a government #ClotShot plot.
This comment is NOTICE of possible criminal liability to Lauren K. Roth and members of the Vaccines and Related Biological Products Advisory Committee who owe duties of care, diligence, good faith, and loyalty in recommending “for” or “against” the EUA amendment for COVID-19 mRNA vaccine in children 6 months through 4 years of age.
Only two deaths are listed herein to establish knowledge. If the amendment is approved, it will have been done by committee members “knowing” of felony crimes in context.
Your investigation of these deaths should include death certificates, autopsy records, witness interviews, and immunization records.
Massachusetts Death Certificate 2022 SFN 5980 is a 7yo girl died January 18, 2022 listed as died from U071 “COVID-19”, B49 “unspecified mycosis”, J450 “predominantly allergic asthma”, and R091 “pleurisy”.
VAERS_ID 2038120 is a 7yo girl in Massachusetts, who received her 2nd dose 1/13/2022 and was reported to VAERS 1/15/2022. PRIOR_VAX states, “Severe nausea and vomiting from 5min post vaccination and for the next 8-10 hours.” SYMPTOM_TEXT states, “Spiked a 103 fever, severe stomachache, has not had a bowel movement since the day before vaccination, which makes today 3 days without one. First vaccine caused severe nausea and vomiting from 5minutes post injection and for the next 8-10 hours.”
This little girl suffered immeasurably 4 to 5 days as her intestines shut down due likely to impeded blood vessels servicing intestines.
Massachusetts Death Certificate 2021 SFN 56611 is a 48yo man died 11/16/2021 listed as died from U071 “COVID-19” and E669 “OBESITY”.
SFN 56611 is known to have died less than 24 hours after inoculation.
In both cases, the Medical Examiners listed the cause of death as “COVID-19”, when it was clearly not COVID-19. And in both cases, the Medical Examiners omitted listing causes Y590 “Viral vaccines“ and T881 “Other complications following immunization, not elsewhere classified”, when these clearly were proximate and actual causes.
Death certificates from the state of Massachusetts are sent to the CDC, a federal entity. Thus, fraud on a state death certificate is a federal crime as it affects federal death records. Several federal felony crimes apply in this instance and are listed below.
If you dismiss this NOTICE and recommend the EUA amendment without first investigating these two deaths, you become liable for inchoate crimes and the felony crime of “misprision of felony.” If a single person subsequently dies as a result of the amendment, all the elements will have been satisfied for you to face felony murder charges or involuntary manslaughter. Qualified immunity is not a valid defense.
18 USC § 4 – Misprision of felony
“Whoever, having knowledge of the actual commission of a felony …, conceals and does not as soon as possible make known the same to some … civil or military authority …, shall be fined under this title or imprisoned not more than three years, or both.”
Felony murder is a homicide that occurs during the commission of an inherently dangerous felony, showing a conscious disregard for human life. A jury decides whether recommending an injection, that you “know” caused death, and that you refused to investigate while “knowing” it caused death, is inherently dangerous.
Here are a few federal statutes likely violated by Medical Examiners in Massachusetts. You are duty-bound to call for investigation of:
- 18 USC § 4 Misprision of felony
- 18 USC § 286 Conspiracy to defraud the government with respect to claims
- 18 USC § 287 False fictitious or fraudulent claims
- 18 USC § 371 Conspiracy to commit offense or to defraud United States
- 18 USC § 1035 False statements relating to health care matters
- 18 USC § 1040 Fraud in connection with major disaster or emergency benefits
There were found sixty likely C19 vaccine deaths in a 25-minute perusal of the 2021 and 2022 death certificates, which extrapolates to hundreds, probably thousands of C19 vaccine deaths in Massachusetts.
Refusal to investigate these fraudulent records is a crime that, because of the felony murder aspect, has no statute of limitations. Five, ten, or twenty years from now, if a federal prosecutor were to learn of this NOTICE, he or she would have significant evidence to bring charges for felony murder.
In summary, this NOTICE places you in a position requiring you to investigate these deaths prior to recommending the amendment. If you dismiss this NOTICE, you may be criminally liable for involuntary manslaughter, felony murder, and a list of federal crimes and inchoate crimes.
Please make the appropriate decision for yourselves and for the children of the United States of America.
Comment Tracking Number
l4d-m52d-ge4m
They Attempt to Justify Approval for Use in Infants and Toddlers
They want the COVID-19 vaccine approval for children so bad, Peter Marks himself and his cronies published the very study he has to use to evaluate for approval.
By James Lyons-Weiler | Popular Rationalism | June 11, 2022
As promised, the FDA has ginned up a report that ostensibly will be used to try to justify “approval” (whatever they mean by that now) of COVID-19 vaccines for infants and toddlers (children < 5 years old). Here’s the report for your reference.
This report comes after a torrent of massive reports from Moderna and Pfizer that claim to review studies of the safety and efficacy of COVID-19 vaccines in children. It is not hard to see what shenanigans the FDA has been up to to try to bolster a vaccine that fewer and fewer adults want. It’s more of the same: exaggerate the apparent risk of the virus and minimizing the perception of risk. In other words, lies.
- There is no evidence of clinical urgency. Infants and toddlers (and children in general) do not get COVID-19; they do not (yet) die from COVID-19. All that can change when antibody dependent enhancement kicks in for the vaccinated. FDA’s own reports cites 1,086 deaths “from COVID-19” and 10,700,000 “cases” of COVID-19 in children aged 0-17. There have been 832 days since April 1, 2020 when diagnoses started for COVID-19. For the entire population of children in the US (73,000,000), the risk of COVID-19 infection since the onset of COVID is 10,700,000/73,000,000 = 0.14657. The risk of a child dying if they have a diagnosis is 1,086/10,700,00 or 1086/10700000 = 0.00010149532. The risk of any child dying of COVID-19 over this time period is 1,086/73000000 = 0.00001487671. The per-day risk is on the order of 1.78806611e-8 (0.000000001788). There is no real unmet clinical need and the FDA needs to go back to college to understand how to use RT-PCR correctly. Children do not get COVID-19, and they do not die.
- Inconsistent use of the idea “vaccinated”. This has been the pattern from the very first study. FDA, CDC, Moderna, Pfizer, and others pull out whatever definition of “vaccinated” they want. Examples: “Vaccinated” is defined in the original trials as people who received both doses and who did not develop COVID-19 before two weeks passed after the second exposure to the vaccine. In fact, that means that people who developed COVID-19 due to disease enhancement were dropped from the study calculations. First, this is the first time people were dropped from a vaccine trial for getting infected with the pathogen targeted by the vaccine up to 13 or 14 days after being vaccinated. Second, it’s actually five entire weeks – one month and one week – 44 days – after the first exposure. ALL of the vaccine efficacy being cited by FDA is suspect. Moderna’s and Pfizer’s vaccines never achieved >90% true vaccine efficacy; the best estimate is more like 75%.
- Inconsistent use of the idea “vaccine efficacy”. Over the time period since the first COVID-19 vaccine trials, various definitions of “vaccine efficacy” have been used. Decreased transmission. Reduction in infection rates. Reduced hospitalization. Presence of neutralizing antibodies. Presence of antibodies. All are used and cited in FDA’s report whenever convenient, all in an ad-hoc manner. It’s more than irritating. It’s moving the goal post and represents reckless (and ineffective) attempts to manipulate public perception. This practice continues in the reports and studies that are cited by FDA. I do not trust the efficacy data FDA cites in their report (why would we given Point 1?).Further evidence of the futility of the evidence used to claim efficacy comes from Moderna’s Sponsor Briefing report to the FDA:“3.3 Regulatory Considerations for Clinical Development of COVID-19 Vaccines in Children
Effectiveness
Regulatory precedent with other preventive vaccines provides a basis for inference of vaccine effectiveness in pediatric populations based on immunobridging to a young adult population in which clinical disease endpoint vaccine efficacy has been demonstrated for the same prototype vaccine. The immune marker(s) used for immunobridging do not need to be scientifically established to predict protection but should be clinically relevant to the disease. Based on available data in humans and animal models, FDA considers neutralizing antibody titers (a functional measure of the vaccine immune response against SARS-CoV-2) to be clinically relevant for immunobridging to infer effectiveness of COVID-19 vaccines in pediatric age groups. Because no specific neutralizing antibody titer has been established to predict protection against COVID-19, two immunogenicity endpoints (GMT and SRR) are considered appropriate for comparing the range of neutralizing antibody responses elicited by the vaccine in pediatric versus young adult populations.
Also embedded in this piece of work is the fact that FDA does not need evidence of long-term immunity; they are settling for something called “immunobridging” – guessing at the efficacy of a vaccine in one clinical population from measurements made from other clinical populaton.
They also are making people dependent on vaccines… expecting patients to have antibodies from one vaccine to the next. This makes no sense immunologically. We don’t need continuously high antibody levels against any pathogen. We have memory B-cells and T-cells. In accepting this paradigm, FDA is completely off its rocker and will cause immune exhaustion with constant vaccinations every 3-4 months.
- Incomplete consideration of the scientific data (Barnstable County, Israel, Ontario). We know that months after vaccination, those who are vaccinated are at higher risk of infection and now of hospitalizations. Data actually show negative vaccine efficacy in children (per Jeremy Hammond). See: “Evidence for Negative COVID-19 Vaccine Effectiveness in Children”. From that article:“vaccine effectiveness (VE) in children becomes(sic) negative within several months since receipt of the second dose.Researchers from the New York State Department of Health published a study on the preprint server medRxiv on February 28 noting that the evidence for vaccine effectiveness in children, particularly those aged five to eleven, was “limited”. So, they aimed to provide data to inform policymaking.“During Omicraon variant predominance,” the authors concluded, “VE against infection declined rapidly” for young children in the state of New York, “with low protection by one month following full-vaccination.”Comparing COVID-19 cases during January between unvaccinated and vaccinated children, they estimated initial vaccine effectiveness for children aged twelve to seventeen to be 76 percent, but this dropped to below 50 percent after just five weeks since receipt of the second dose.Moreover, for young children (aged five to eleven), they observed a drop from 65 percent to just 12 percent after only one month.Thereafter, their estimate indicated significantly negative effectiveness for this age group, as shown in Figure 2 of their paper: by 35 to 41 days, VE reached negative 10 percent, and by 42 to 48 days, it reached negative 41 percent.

Jeremy goes on to report (correctly) that the authors of the article misinterpreted their own data. History will remember Jeremy as a reporter with great integrity.
- Moderna and Pfizer reports fail to study long-term risks. Like I said, more of the same shenanigans. In this report, for example, Moderna offers data on myocarditis only up to Day 28 after the vaccine. Why Day 28? Why not “since the vaccine has been administered” to more accurately reflect the real-world clinical situation? They also state that myocarditis in a large concern in people infected with SARS-CoV-2 – but the comparison is to the uninfected, not the vaccinated, and we know that the spike protein is the cause (syncytia among heart muscles caused by the spike protein). The spike protein, of course, is the basis of their mRNA vaccines.
- Incestuous COIs/Unjustified Influence by Regulators. Peter Marks is charged with setting the decisions at FDA whether to consider vaccines for specific populations. Why the hell is he involved in a study conducted to bolster the vaccines he is going to have to decide upon? See “Benefit-risk assessment of COVID-19 vaccine, mRNA (Comirnaty) for age 16–29 years”. That “study” is also guilty of all of the same loose logic as above; it is noteworthy that the study assumes as “worst case scenario” of zero deaths from myocarditis following COVID-19 vaccination (Credit: Toby McDonald, who wrote this to me:“I’m reading the Moderna “Sponsor Briefing Document” and they built their benefit-risk assessment off of Funk et al. (2022). So I looked up Funk and it’s a recent paper by six staffers at the FDA including Peter Marks, Richard Forshee, and Hong Yang (who wrote the dreadful benefit-risk assessment for kids 5 to 11 back in October). Quite literally in their “worst-case scenario” they predict 0 deaths from myocarditis in the vaccine group. It’s a stunning work of fiction.”
- I’m on an email thread with Steve Kirsch (he considers me part of his “debate team”. Last week, Steve challenged Peter Marks to a debate:“Hi Peter,You are right about the vaccine uptake problem. According to independent survey we just commissioned, only 33% of Americans opted to go further than the first 2 doses.You were quoted in that CNN article:“We do have a problem with vaccine uptake that is very serious in the United States and anything we can do to get people more comfortable to be able to accept these potentially life-saving medical products is something that we feel we are compelled to do,” said Dr. Peter Marks, director of the Center for Biologics Evaluation and Research.Isn’t it time for you to end the misinformation problem by debating us in a public forum?My colleagues and I look forward to hearing from you.
The only way to end the misinformation is to debate the top misinformation spreaders. You will never win by trying to censor us.
We would be HAPPY to debate to you to end the misinformation problem. As you can see from this slide deck, all the evidence we’ve been able to find shows there was clinical trial fraud and that the vaccines are very dangerous. We would love to know how we got it wrong
I look forward to hearing from you.
-steve
To my knowledge, Marks has not replied. I replied to Steve and the entire email thread, including Marks, though:
“Steve,
History is going to remember one person on this email thread in a manner in which I would not ever care to be seen associating with.
I would therefore decline to participate in such a debate.
Sincerely,
James Lyons-Weiler, PhD

I could continue and debate dozens more points in the report dump by the FDA. I don’t have to. Marks himself provides evidence of being way off-target immunologically and lying about the “need” for COVID-19 vaccines for children.
Here’s an old video of Prevaricating Peter lying about the need for “high antibody titres” for immunity, and that children’s immune response is “not enough for some of these variants” (no data on that, just words):
The comments in that video have not aged well. Call your Senator and Congressional Reps and demand that Peter Marks resign. Email them this article. Marks and the FDA are NOT basing their considerations on independent fact, science and logic. He and his cronies are either incompetent or working for the industry. Either way, he and his cronies have to go.
Doctors Sue FDA, Allege Crusade Against Ivermectin ‘Unlawfully Interfered’ With Their Ability to Treat Patients
The Defender | June 6, 2022
Three physicians are suing the U.S. Food and Drug Administration (FDA) for launching what they allege is a “crusade” against ivermectin as a treatment for COVID-19 that “unlawfully interfered” with the doctors’ ability to practice medicine.
In a lawsuit filed June 2, Drs. Robert L. Apter, Mary Talley Bowden and Paul E. Marik argued the FDA acted outside of its authority by directing the public, including health professionals and patients, to not use ivermectin — even though the drug is fully approved by the FDA for human use.
The suit, filed in the U.S. District Court, Southern District of Texas, Galveston Division, also names the U.S. Department of Health and Human Services (HHS), HHS Secretary Xavier Becerra and Robert Califf, acting FDA commissioner.
According to the complaint:
“The FDA generally cannot ban particular uses of human drugs once they are otherwise approved and admitted to the market, even if such use differs from the labeling — commonly referred to as ‘off-label’ use.
“The FDA also can not advise whether a patient should take an approved drug for a particular purpose. Those decisions fall within the scope of the doctor-patient relationship.
“Attempts by the FDA to influence or intervene in the doctor-patient relationship amount to interference with the practice of medicine, the regulation of which is — and always has been — reserved to states.”
The plaintiffs said their lawsuit isn’t about whether ivermectin is an effective treatment for COVID-19. It’s about who determines the appropriate treatment for each unique patient and whether the FDA can interfere with that process.
In their complaint, they site an FDA publication, “Why You Should Not Use Ivermectin to Treat or Prevent COVID-19,” and tweets from the FDA — including one implying that ivermectin is intended only for animals — among examples of the FDA discouraging the use of ivermectin.
The plaintiffs also argued if the FDA is allowed to interfere with the practice of medicine now, using the pandemic as a cover, “this interference will metastasize to other circumstances, destroying the carefully constructed statutory wall between federal and state regulatory powers, and between the FDA and the professional judgment of health professionals.”
“This lawsuit, brought by three eminently qualified physicians, is a welcome development,” said Mary Holland, Children’s Health Defense president and general counsel.
Holland told The Defender :
“These doctors rightfully assert that the FDA, assisted by corporate media, have unlawfully interfered in the doctor-patient relationship and the appropriate treatment for individual patients. Regulating the doctor-patient relationship is an area of well-established state, not federal, law.
“I hope these plaintiffs will enjoin the FDA from continuing to restrict access to ivermectin and from penalizing healthcare practitioners who use this licensed drug for their patients.”
The plaintiffs: well-respected in their field, high success rate treating COVID patients
Apter, who is licensed to practice medicine in Arizona and Washington and has a COVID-19 patient survival rate of more than 99.98%, was referred to the Washington Medical Commission and Arizona Medical Board for disciplinary proceedings for prescribing ivermectin to treat COVID-19.
In a press release, Apter said, “If doctors are freed to treat patients according to their best judgment and unprejudiced evaluation of the medical literature, many thousands more deaths and serious disabilities will be averted.”
Apter said the FDA’s pronouncements against the use of ivermectin “have been the basis for disciplinary actions against doctors, interfere with the doctor-patient relationship, and have had a severe chilling effect on the use of life-saving medication for a deadly disease.”
In the lawsuit, Apter argued that government pressure, “largely through the FDA,” also led pharmacies — especially in large corporate chains — to refuse to fill ivermectin prescriptions for COVID-19, because that position is supported by the FDA.
Bowden, who according to the lawsuit has 40 years of experience in emergency medicine, began recommending ivermectin to treat COVID-19 in early 2020. She treated more than 3,900 patients for COVID-19, with a success rate of over 99.97%.
She said the FDA’s actions regarding ivermectin, specifically its directives to stop using the drug to treat COVID-19, harmed Bowden’s ability to practice medicine and treat patients.
Bowden’s employer, Houston Methodist Hospital, last year forced her to resign by suspending her privileges for spreading “COVID misinformation.”
Bowden said she is “fighting back — the public needs to understand what the FDA has done is illegal, and I hope this suit will prevent them from continuing to interfere in the doctor-patient relationship.”
In an interview earlier this year with The Defender, Bowden said she was all for the COVID vaccines when they first came out — it was only when she started seeing what was happening with all the breakthrough cases that she wondered, “Why am I seeing so many COVID cases among the fully vaccinated?”
Then her patients began having adverse reactions. “If I hadn’t seen that firsthand, I would still think the vaccine was the way to go,” she said.
As the pandemic evolved, Bowden developed protocols for preventing and treating COVID patients. She said she’s seen excellent results.
“The basis of it is ivermectin,” she said. “And also vitamins C and D, quercetin and zinc, and black seed oil. It’s nothing complicated — and it’s just like with anything in medicine — not one size fits all — protocols are guidelines.”
The controversy over prescribing ivermectin was initially “intimidating and isolating,” she said. “I thought I was a little bitty island in a huge ocean, and now I realize that I’m part of at least half a continent.”
Marik, author of more than 750 publications, was professor of medicine and chief of pulmonary and critical care medicine at Eastern Virginia Medical School (EVMS) in Norfolk, Virginia, from 2009 through 2021. He also served as a director of the intensive care unit at Sentara Norfolk General Hospital.
He developed a protocol for EVMS for treating COVID-19, called the EVMS COVID-19 Management Protocol, which included the MATH+ Protocol.
However, according to the lawsuit, Marik was forced to resign from his positions at EVMS and Sentara Norfolk General Hospital for promoting the use of ivermectin — “as well as other safe, cheap, and effective off-label FDA-approved drugs” — to treat COVID-19 following the FDA’s attempts to stop use of those drugs for that purpose.
Marik alleged in the lawsuit that refusing to allow patients to receive effective early treatment for COVID-19 “led to innumerable hospitalizations and deaths, and caused extreme distress for patients, their families, and health professionals.”
Boyden, Gray & Associates, a Washington, DC-based law firm, is representing the plaintiffs.
Ivermectin was developed in the 1970s as a veterinary medicine to treat parasitic diseases in livestock, but a decade or so later was hailed as a “wonder drug” and received approval for human use as a therapeutic against diseases such as river blindness — or onchocerciasis — and lymphatic filariasis, according to Newsmax.
Since 1987, it has been used safely in 3.7 billion doses worldwide. William Campbell and Satoshi Omura won the 2015 Nobel Prize in Physiology or Medicine for their research on the drug.
Studies show ivermectin is associated with lower COVID-19 death rates, but the FDA — with help from mainstream media — continues to state the drug is ineffective for treating COVID.
© 2022 Children’s Health Defense, Inc. This work is reproduced and distributed with the permission of Children’s Health Defense, Inc. Want to learn more from Children’s Health Defense? Sign up for free news and updates from Robert F. Kennedy, Jr. and the Children’s Health Defense. Your donation will help to support us in our efforts.
Ghost Shot: Pfizer quietly admits it will never manufacture original FDA approved COVID vaccines
Company claims it is manufacturing Comirnaty product with new formula

By Jordan Schachtel | The Dossier | June 3, 2022
The August 23, 2021 FDA approval of Pfizer’s Comirnaty vaccine was a cause for celebration. Marked as a turning point in the battle against COVID19, the announcement was highly publicized by the Biden Administration with the clear intention to extinguish “vaccine hesitancy” and boost uptake.
It was celebrated as a cause for national relief, and many Americans arrived at their local pharmacies under the impression, via government and pharmaceutical propaganda, that they were receiving an FDA-approved COVID vaccine. Yet that legally distinct product, as we know it, never existed. And now we know, via Pfizer, that it will never exist.
For the uninitiated:
Comirnaty is a legally distinct product from the emergency use authorization (EUA) shots, and It has never made its way to market. For months on end, no such vaccine has ever become available. Those who received the “Pfizer shot(s)” have been injected with the emergency use authorization (EUA) version of the shots. See my piece in The Dossier for more info:
Shell Game? There remains no FDA approved COVID vaccine in the United States
The information operation succeeded. There was indeed an FDA approved vaccine, at least on paper, but you couldn’t get it.
When originally confronted with this ordeal, Pfizer labeled this issue an inventory question that had nothing to do with the legal distinction between an experimental EUA product and an FDA-approved vaccine. Up until just weeks ago, this was the statement up on the CDC website via Pfizer:
“Pfizer received FDA BLA license on 8/23/2021 for its COVID-19 vaccine for use in individuals 16 and older (COMIRNATY). At that time, the FDA published a BLA package insert that included the approved new COVID-19 vaccine tradename COMIRNATY and listed 2 new NDCs (0069-1000-03, 0069-1000-02) and images of labels with the new tradename.
At present, Pfizer does not plan to produce any product with these new NDCs and labels over the next few months while EUA authorized product is still available and being made available for U.S. distribution. As such, the CDC, AMA, and drug compendia may not publish these new codes until Pfizer has determined when the product will be produced with the BLA labels.”
In May, Pfizer updated its statement to mention a December 2021 licensed Comirnaty product, which was granted a license four months after the highly-publicized August FDA press release.
And just last week, Pfizer finally acknowledged that its original licensed product will never be distributed. In an unreported update on the CDC website, Pfizer told the agency:
“Pfizer received initial FDA BLA license on 8/23/2021 for its COVID-19 vaccine for use in individuals 16 and older (COMIRNATY). At that time, the FDA published a BLA package insert that included the approved new COVID-19 vaccine tradename COMIRNATY and listed 2 new NDCs (0069-1000-03, 0069-1000-02) and images of labels with the new tradename. These NDCs will not be manufactured. Only NDCs for the subsequently BLA approved tris-sucrose formulation will be produced.”
The key distinction between the originally approved formulation and the tris-sucrose formulation is that — according to manufacturers — the latter can be held for a much longer period of time outside of an ultra cold freezer. These freezers cost over $10,000 a piece and each unit uses as much energy per day as an average American household. Improper storage can render the mRNA unstable.
Notably, the clinical trials for the Pfizer shot were conducted without the modified tris-sucrose ingredient. Given the partisan nature of Pfizer, the corporate media, government health bureaucracies, and your correspondent’s lack of expertise in this area, it is unclear whether this is significant.
Another notable thing to look out for in the coming days and weeks is the possibility that the subsequently FDA approved product finally becomes available in the United States. In recent days, the CDC removed the language of “not orderable at this time” above the description of both Comirnaty and Moderna’s Spikevax.
Additionally, as reported by Uncover DC, the Defense Department appears to be in the early stages of ordering what it has interpreted as a legally required minimum of Comirnaty in order to continue its mRNA mandate of American service members.
The FDA’s proposed “Future Framework” is the worst idea in the history of public health
If approved on June 28, all reformulated Covid-19 shots will skip clinical trials
By Toby Rogers | May 31, 2022
I. Pfizer and Moderna’s Dilemma
Pfizer and Moderna have a problem — their Covid-19 shots do NOT work. Everyone knows this. The shots do not stop infection, transmission, hospitalization, nor death. Over half a billion doses of this product have been injected into Americans in the past 17 months and these shots have made NO discernible impact on the course of the pandemic. Far more Americans have died of coronavirus since the introduction of the shots than before they were introduced.
Pfizer and Moderna are making $50 billion a year on these shots and they want that to continue. So they need to reformulate the shots. Maybe target a new variant, maybe change some of the ingredients — who knows, these shots don’t work so it’s not clear what it will take to get them to work. This is a problem because reformulated shots mean new clinical trials and new regulatory review by the FDA. There is a decent chance that any reformulated shot might fail a new clinical trial and the public is deeply skeptical of these shots so the scrutiny would be intense.
So Pfizer and Moderna have figured out a way to use regulatory capture to get their reformulated Covid-19 shots approved WITHOUT further clinical trials. Their scheme is called the “Future Framework” and it will be voted on by the FDA’s Vaccines and Related Biological Products Advisory Committee (VRBPAC) on June 28.
II. Doubling down on a failed strategy
Viruses vary by region. At any given time, the influenza strain circulating in England is different than it is in South Africa which is different than in southeast Asia. However, pharmaceutical companies prefer to create one-size-fits-all vaccines in order to decrease manufacturing costs and thereby increase profits. So the W.H.O. and public health agencies around the world (including FDA and CDC) have created a vast “influenza surveillance network” that identifies the different influenza strains in circulation. Then they engage in an elaborate theatrical performance called the “flu strain selection process” where they select four influenza strains that will go into the one-size-fits-all flu vaccine used throughout the world that year.
This carefully choreographed process is a complete and total failure. This is not a surprise — using a one-vaccine-fits-all approach to prevent a rapidly evolving virus that varies by region is never going to work. Lisa Grohskopf from the CDC’s Influenza Division reports that last year the flu shot was somewhere between 8% and 14% effective (based on data from seven sites that participate in the U.S. Flu Vaccine Effectiveness Network).

But a case study of a flu outbreak at the University of Michigan between October and November 2021 found that the effectiveness of the flu vaccine was literally zero.
![]()
Over the last thirty years, the federal government has paid out more compensation for adverse events in connection with the flu shot than any other vaccine — so we know that the shot comes with a high rate of harms. Given that the flu shot does not stop the flu, the harms thus outweigh the benefits.
In a sane world, the WHO, FDA, and CDC would admit that they made a strategic mistake and then change course to find better ways to support the human immune system. But we don’t live in a sane world. Instead, the FDA is proposing to take the failed flu strain selection process and apply it to future Covid-19 shots.
III. The FDA knew that Covid-19 shots would fail but they proceeded anyway
There are a quadrillion x quadrillion viruses in the world (literally more viruses on earth than stars in the known universe). Only a couple hundred of those seem to have the potential to impact human health. But some viruses make better candidates for a vaccine than others. Viruses that have been around a long time, that are very stable and evolve slowly are the best candidates for a vaccine.
Viruses that evolve rapidly are bad candidates for a vaccine. There is no vaccine for the common cold nor HIV because these viruses evolve too quickly. The SARS-CoV-2 virus is a bad candidate for a vaccine which is why all previous attempts to develop a vaccine against coronaviruses have failed (they never made it out of animal trials because all of the animals died during challenge trials or were injured by the vaccine).
What are some of the bad things that can happen when you vaccinate against a rapidly evolving virus? Original antigenic sin, antibody dependent enhancement, and the possibility of accelerating the evolution of the virus in ways that make it more virulent (and even more resistant to vaccination).
Trevor Bedford has his own lab at the Fred Hutchinson Cancer Center where he researches the evolution of Covid-19. He gave a fascinating presentation at the April 6 meeting of the FDA’s Vaccines and Related Biological Products Advisory Committee meeting where he explained that SARS-CoV-2 is evolving rapidly. He explained that SARS-CoV-2 evolves twice to ten times as fast as the flu virus and these mutations “substantially” reduce vaccine effectiveness. Following the introduction of Covid-19 vaccines, the evolution of the virus has accelerated.
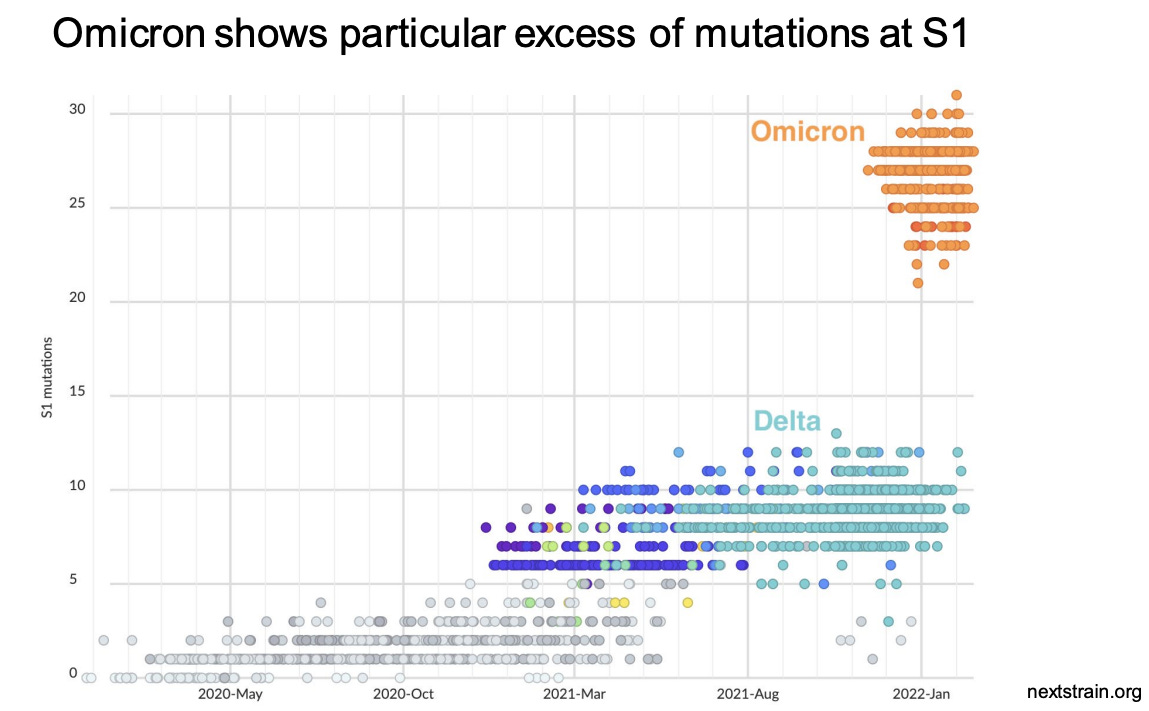
Dr. Bedford’s presentation rattled some of the smarter members of the VRBPAC because his data scream — “SARS-CoV-2 is a bad candidate for a vaccine!” But FDA officials just mumbled some platitudes and then continued on with the meeting.
The only way out of the pandemic is to withdraw these vaccines from the market and pivot to therapeutics. Instead, the FDA is proposing to just hide the data from the American people.
IV. The “Future Framework” = no more clinical trials for Covid-19 shots ever again
The purpose of the “Future Framework” is to rig the Covid-19 vaccine regulatory process in perpetuity in favor of the pharmaceutical industry. If this “Future Framework” is approved all future Covid-19 shots, regardless of the formulation, will automatically be deemed “safe and effective” without additional clinical trials because they are considered “biologically similar” to existing shots.
This is literally the worst idea in the history of public health.
If you change a single molecule of mRNA in these shots it will change health outcomes in ways that no one can anticipate. That necessarily requires new clinical trials — which is what the FDA is proposing to skip.
The FDA’s “expert advisory committee” (VRBPAC) met on April 6, 2022 to discuss the “Future Framework” for the first time. All of the committee members agreed that Covid-19 shots are not working, that boosting multiple times a year was not feasible, and that the shots need to be reformulated. They also unanimously agreed that there are no “correlates of protection” that one can use to predict what antibody levels would be sufficient to prevent SARS-CoV-2 infection.
On June 28 the VRBPAC will meet once again to discuss the “Future Framework” and it will be presented as a done deal because manufacturers want a decision on vaccine strain selection by June in order to deliver shots for autumn vaccination appointments.
So if the FDA authorizes Covid-19 shots for kids on June 14 and 15 and then approves the “Future Framework” on June 28th, the shots that will be given to kids in the fall will be the reformulated shots that skipped clinical trials.
V. Monovalent Covid-19 shots failed, so maybe throwing two, three, or four variants into a single shot will make it better?
When it comes to the flu shot, the FDA tries to hedge their bets by putting four strains of the virus into a single shot (so called “quadrivalent” vaccines). As I explained above, this strategy does not work. But these people are not very clever so that’s exactly what they are planning to do with future Covid-19 shots.
Moderna is already signaling that they intend to manufacture a Covid-19 shot with the Alpha variant and then, to make it “new and improved (TM)”, they will add genetically modified mRNA targeting the Beta variant. Here’s the best part — Moderna claims that this formulation (Alpha + Beta) will somehow protect against Omicron variants — even though by the time these reformulated shots get to market, none of these variants will likely still be in widespread circulation.
There are reasons to believe that this approach will make future Covid-19 shots even less effective and more dangerous than the current failed Covid-19 shots.
Think about it. The more mRNA you put into a shot, the higher the adverse event rate (as the genetically modified mRNA hijacks the cell and starts cranking out spike proteins). So if Pfizer and Moderna put more mRNA into these shots (in order to cover multiple variants) adverse event rates will skyrocket.
But if Pfizer and Moderna put less mRNA per variant into a shot (in order to keep the total amount of mRNA at 100 mcg for Moderna and 30 mcg for Pfizer) then the effectiveness against any one particular variant will be reduced.
The Future Framework is 100% guaranteed to fail. If the “Future Framework” is approved, effectiveness of these shots will decrease, adverse events will increase, these shots will fuel the evolution of variants that evade the vaccines, and there will be no clinical trial data before these reformulated Covid-19 shots are unleashed on the unsuspecting public.
VI. Summary
The FDA’s Vaccines and Related Biological Products Advisory Committee will meet on June 28 to vote on a “Future Framework” for evaluating so-called “next generation” Covid-19 shots. The “Future Framework” is a plan to rig the Covid-19 vaccine regulatory process in perpetuity.
The “Future Framework” would take the “flu strain selection process” that fails every year and apply it to future (reformulated) Covid-19 shots. Federal bureaucrats, many of whom have financial conflicts of interests, would choose which SARS-CoV-2 variants to include in a yearly (or twice yearly) Covid-19 shot. In the process, all future Covid-19 shots will be deemed automatically “safe and effective” without further clinical trials because they are considered “biologically similar” to existing Covid-19 shots.
The “Future Framework” is the most reckless idea in the history of public health. It shows that the FDA has completely abandoned science and its statutory duty to protect the public. If the Republic is to survive, we must stop the “Future Framework” before June 28.
VII. Call to action
We have very little time and an enormous challenge in knocking this proposal down before the VRBPAC meets on June 28. So I am asking to you to contact your elected officials to tell them to reject this dangerous proposal.
Below are talking points that you can paste into an email, a script that you can use on the phone, and a tool for looking up your elected officials. I am only asking you to contact 8 officials — the President and Vice President; your two Senators and U.S. Representative; and your Governor, state House/Assembly member, and state Senator. Please be respectful but make it clear that this plan must be stopped.
Talking points (to paste into an email, letter, or fax)
Subject line: NO “flu framework” for future Covid-19 shots
The FDA and CDC are developing a “Future Framework” to authorize future Covid-19 shots without requiring additional clinical trials. This would be a public health disaster. I am asking you to contact the FDA to tell them to stop all work on this “Future Framework” immediately. If the FDA proceeds with this “Future Framework” I am asking you to eliminate all funding for the FDA in this year’s budget.
Phone script
Hi, my name is ____________. I live at __________________[address]. I’m calling because the FDA is proposing a “Future Framework” to authorize future Covid-19 shots without requiring additional clinical trials. This would be a public health disaster. I am asking you to contact the FDA to tell them to stop all work on this “Future Framework”. If the FDA proceeds with this “Future Framework”, I am asking you to eliminate all funding for the FDA in this year’s budget.
Whom to contact: 8 phones calls, letters, emails, or faxes:
President Joseph R. Biden
The White House
1600 Pennsylvania Ave NW
Washington, DC 20500
(202) 456-1111 (The White House comment line is open between the hours of 11 to 3 p.m. EST Tues.-Thurs.)
https://www.whitehouse.gov/contact/
https://twitter.com/POTUS
Vice President Kamala Harris
The White House
1600 Pennsylvania Ave NW
Washington, DC 20500
(202) 456-1111 (between the hours of 11 to 3 p.m. EST Tues.-Thurs.)
https://www.whitehouse.gov/contact/
https://twitter.com/VP
You can look up contact info for your two U.S. Senators and U.S. Representative here:
https://www.govtrack.us/congress/members/map
The message for State elected officials is slightly different:
Hi, my name is ____________. I live at __________________[address]. I’m calling because the FDA is proposing a “Future Framework” to authorize future Covid-19 shots without requiring additional clinical trials. This would be a public health disaster. If the FDA proceeds with this “Future Framework” I are asking you to nullify the actions of the FDA and reject any Covid-19 shots that have not gone through proper clinical trials.
This is a great tool to look up contact info for your Governor, state Senator, and state House/Assembly member:
That’s it, just 8 people. We want to let them know that we are watching, that we understand what they are up to, and that this wretched plan must be stopped.
Extra credit:
Here are the email addresses for all of the public health political appointees, FDA staff, and VRBPAC members who have a say in connection with the “Future Framework”. Let’s contact them as well (proposed subject line and email text below).
Subject line: The “Future Framework” is the WORST idea in the history of public health. Please vote NO.
1. The FDA must revoke the authorizations for Moderna, Pfizer, and J&J Covid-19 shots and withdraw them from the market immediately. SARS-CoV-2 was never a good candidate for a vaccine. These shots do not stop infection, transmission, hospitalization, nor death. They appear to have negative efficacy and are driving the evolution of variants that evade vaccines. The pandemic will never stop as long as the FDA and CDC are promoting shots that lack sterilizing immunity.
2. The FDA and CDC must pivot to therapeutics. This was always the answer. About twenty off-the-shelf treatments are more effective than vaccines (if used for prophylaxis or early intervention). Get these safe and effective medicines to people who need them and let doctors be doctors again and treat patients based on their own best clinical judgment.
3. Any reformulated Covid-19 shots MUST go through proper clinical trials and FDA review. That means:
• Large (50,000+ person) double-blind randomized controlled trials with inert saline placebos conducted by an independent third party;
• Safety and efficacy studies for two years prior to any application; the treatment and control groups must be followed for 20 years to monitor adverse events and all-cause mortality (no more wiping out the control group after 6 months to hide bad outcomes);
• Greater than 90% efficacy with less than 1% Grade 3 Adverse Events; and
• Proper monitoring for carcinogenesis, mutagenesis, and impairment of fertility.
sean.mccluskie@hhs.gov, commissioner@fda.hhs.gov, ashish.jha@whitehouse.gov, Aux7@cdc.gov, Peter.Marks@fda.hhs.gov, Hong.Yang@fda.hhs.gov,
Richard.Forshee@fda.hhs.gov, Huilee.Wong@fda.hhs.gov, Leslie.Ball@fda.hhs.gov, Doran.Fink@fda.hhs.gov, hanae@bcm.edu, paula.annunziato@merck.com,
adam.berger@nih.gov, hbernstein@northwell.edu, acohn@cdc.gov, anc0@cdc.gov, hjanes@fredhutch.org, hgans@stanford.edu, david.kim@hhs.gov,
asmonto@umich.edu, offit@chop.edu, spergam@fredhutch.org, Jportnoy@cmh.edu, erubin@hsph.harvard.edu, erubin@nejm.org, ashane@emory.edu,
swamy002@mc.duke.edu, fullerao@umich.edu, RandyHawkins@cdrewu.edu, officeofthepresident@mmc.edu, JYLee@uams.edu,
ofer.levy@childrens.harvard.edu, wayne_marasco@dfci.harvard.edu, cmeissner@tuftsmedicalcenter.org, mrn8d@virginia.edu,
FDA announces updated schedule for the June meetings regarding five pivotal vaccine decisions
Who needs data when you’ve got regulatory capture?
By Toby Rogers | May 29, 2022
I. The June FDA meetings
This week the Washington Post copied and pasted from a Pfizer press release to announce yet another scientific miracle(TM) that will completely fail in practice. In the process WaPo also got some quotes from the FDA who have now nailed down the schedule for the 4 meetings in June in which they intend to assemble the final pieces for Pharma’s permanent dominance over the American people.
The new schedule is as follows:
June 7, Novavax
June 14, Moderna in kids 6 to 17 years old
June 15, Moderna in kids 6 months to 5 years AND Pfizer in kids 6 months to 4 years
June 28, “Future Framework” (the plan to skip clinical trials in perpetuity)
There is a lot to parse in the WaPo’s brief article.
Contrary to the breathless headline, they still don’t have any data.
Pfizer and BioNTech said the 80 percent efficacy finding was preliminary and based on 10 cases of Covid-19 in the study population as of the end of April. Once 21 cases have occurred, the companies will conduct a more formal analysis of efficacy… Pfizer and BioNTech said they plan to finish filing data with the FDA this week — and warned that the efficacy number was fluid because results are still arriving.
Let’s recap how we got here:
🚩 The Pfizer clinical trial in kids under 5 failed in December 2021.
🚩 So Pfizer added a third dose and that trial also apparently failed in February (which is why Pfizer was forced to withdraw its application on February 10).
🚩 Now Pfizer is describing a jerry-rigged trial of a third dose in 1,678 kids ages 6 months to four years old. Pfizer did not disclose how the kids were divided between the treatment and control group so it is impossible to run our own calculations on efficacy. Out of that sample, 10 developed Covid — although it is not clear how the 10 were distributed between the treatment and control group. (I suppose some quant on Twitter will figure out how to work backwards from Pfizer’s claims to calculate the numbers in each of these categories but needless to say, this is not the proper way to do science.) Of course Pfizer also failed to describe the contents of the “placebo.”
How exactly will Pfizer double the number of Covid-19 cases in the clinical trial in the next month given that 74.2% of kids already had natural immunity in February which means that nearly 100% of children likely have natural immunity by now?
Also, is the FDA seriously considering basing national policy, that impacts 18 million children, by relying a study with only 10 cases? It appears that the FDA is not even pretending to care about science anymore.
What little data they have will be based on antibodies in the blood, not health outcomes in the real world. That’s strange because the members of the FDA’s Vaccines and Related Biological Products Advisory Committee unanimously acknowledged on April 6 that there are “no correlates of protection” in connection with Covid-19 shots (this means that there are no valid proxy measures, such as antibody counts, that can determine whether someone who has received this shot is immune to the virus or not.)
WaPo continues:
While the adult trials recruited tens of thousands of volunteers and waited to see if vaccinated people were better protected, the children’s vaccine trials were primarily designed to measure immune responses using blood tests.
No they were not “primarily designed to measure immune responses using blood tests.” The studies were intentionally undersized to hide harms from the shots in addition to other tricks that they use to skew the results (such as kicking you out of the trial if you call 911 or go the the emergency room). But when one shrinks the sample size, surprise! it becomes impossible to detect actual health benefits from the shots (the signal would have been tiny if at all, but when one uses a sample that small then any positive signal can also disappear into statistical insignificance.)
II. The bigger picture
Tony Fauci and the NIAID funded the creation of a chimera virus that escaped a bioweapons lab and killed 6.3 million people worldwide.
Public health authorities have blocked access to safe and effective prophylaxis and early treatment throughout the pandemic in order to create the market for Covid-19 vaccines.
Covid-19 shots skipped essential safety steps (e.g. challenge trials in animals) and were rushed to market with no long term data.
In practice the mRNA shots suppress immune function for 6 weeks after the first shot, provide about two months worth of protection against coronavirus, then efficacy wanes quickly and becomes negative after six months. Meanwhile, these shots cause more side effects than any vaccine ever invented.
Popular support for the current regime has collapsed. More people have died of Covid under Mr. I Believe the Science(TM) than under Orange Man Bad. Only hypochondriacs in blue states seek out additional doses. Meanwhile Sudden Adult Death Syndrome stalks the true believers. In the past 48 hours alone actor Ray Liotta, Andy Fletcher of Depeche Mode, British drummer Alan White (from the band YES), and comedian Phil Butler were all likely killed by Covid-19 shots. It’s impossible to hide all of the bodies at this point.
The FDA seems to know that their window is closing to implement the Final Solution. So they are rushing to put the finishing touches on their plans to inject this toxic junk into the littlest kids in America. The FDA knows that these shots cannot pass proper regulatory review so they’ve developed a plan to rig the process in favor of Pharma in perpetuity. On June 28, the FDA’s “expert advisory committee” will vote on a “Future Framework” whereby all future (reformulated) Covid-19 shots will automatically be deemed “safe and effective(TM)” without any additional clinical trials, on the theory that they are “biologically similar” to existing Covid-19 shots.
What this means is that by fall, the Covid-19 shots that they will be injecting into Americans of all ages will have a new formula that skipped clinical trials altogether.
Injecting people with genetically modified mRNA that skipped clinical trials is genocide. It’s slower than the Nazi Final Solution. But it’s genocide all the same. Indeed the slower pace of the FDA Final Solution (5% to 15% increase in all cause mortality every year) might be even more lethal in the long run. It’s sinister in that they are intentionally building in plausible deniability (‘the FDA said it was safe’) to help the medical establishment feel virtuous while participating in genocide.
I’ll just conclude by saying: be careful what you wish for FDA. The tide has already turned. The American people know exactly what you are doing. We have the receipts. It will be relatively easy to secure a conviction at Nuremberg 2.0 — we literally have you on video committing crimes against humanity. As a reminder, the courts have determined that “I was just following orders” is not a valid defense.
FDA Dumps More Pfizer Documents: Why Were So Many Adverse Events Reported as ‘Unrelated’ to Vaccine?
By Michael Nevradakis, Ph.D. | The Defender | May 17, 2022
The latest release of Pfizer-BioNTech COVID-19 vaccine documents raises questions about how frequently adverse events experienced by clinical trial participants were reported as “unrelated” to the vaccine.
The 80,000-page document cache released May 2 by the U.S. Food and Drug Administration (FDA) includes an extensive set of Case Report Forms (CRFs) from Pfizer trials conducted at various locations in the U.S.
The documents also include the “third interim report” from BioNTech’s trials conducted in Germany (accompanied by a synopsis of this report and a database of adverse events from this particular set of trials).
The FDA released the documents, which pertain to the Emergency Use Authorization (EUA) of the vaccine, as part of a court-ordered disclosure schedule stemming from an expedited Freedom of Information Act (FOIA) request filed in August 2021.
Public Health and Medical Professionals for Transparency, a group of doctors and public health professionals, submitted the FOIA request.
Adverse events during Pfizer vaccine trials in the U.S. usually reported as ‘unrelated’ to vaccination
Pfizer conducted a series of vaccine trials at various locations in the U.S., including the New York University Langone Health Center, Rochester Clinical Research and Rochester General Hospital (Rochester, New York) and the J. Lewis Research, Inc. Foothill Family Clinic (Salt Lake City, Utah).
The Pfizer documents released this month by the FDA included a series of CRFs for patients who suffered some type of adverse event during their participation in the COVID-19 vaccine trials.
As the documents reveal, despite the occurrence of a wide range of symptoms, including serious cardiovascular events, almost none were identified as being “related” to the vaccine.
Such serious yet “unrelated” adverse events included:
- Acute asthma exacerbation
- Aortic aneurysm (listed as a pre-existing condition)
- Appendicitis (requiring hospitalization)
- Atrial defibrillation
- Cardiac arrest and acute respiratory failure, requiring hospitalization, sustained by a patient who then was “lost” (could not be located for continued participation in the trial)
- Chest pain (requiring hospitalization, later listed as cardiac ischemia)
- Coronary artery occlusion (listed as both serious and life-threatening)
- Injuries sustained from a fall
- Intermittent non-cardiac chest pain (requiring hospitalization)
- Left breast cancer (listed as a pre-existing occult malignancy)
- Neuritis (peripheral nerve Injury), listed as “unrelated” to the vaccine but related to the blood draw during vaccination
- Pulmonary embolism and bilateral deep venous thrombosis
- Respiratory failure (requiring hospitalization)
- Right ureteropelvic junction obstruction (requiring hospitalization, listed as congenital)
- Small bowel obstruction, listed as “unplanned,” and a panic attack
Of the CRFs found in the documents released this month, only one adverse event is clearly specified as being related to the vaccination: a participant who suffered from psoriatic arthritis, with no prior history of the condition.
In addition, several CRFs indicated exposure during pregnancy (see here and here), or during a partner’s pregnancy (see here and here). However, the documents provided do not appear to have provided any follow-ups regarding any outcomes or potential adverse events for the participants, their partners or their newborn babies once born.
In some instances, while the CRFs claimed the adverse events suffered by patients were not related to the vaccine, their cause was unspecified, simply indicated as “other,” while in another case, a participant’s “unplanned” small bowel obstruction and panic attacks were listed as being unrelated to the vaccination despite no relevant medical history pertaining to the SAEs (severe adverse events) in question.
Did Pfizer hide critical information from regulators?
It is difficult to draw concrete conclusions about any specific case from the data provided by CRFs and vaccine trial summaries.
However, what raises eyebrows is the very large number of adverse events — often serious and often requiring the hospitalization of the patients involved — that were determined to be “unrelated” to the administration of the COVID vaccine.
Previously released Pfizer documents also included discrepancies in the recording of adverse events.
According to investigative journalist Sonia Elijah, these discrepancies include:
- Trial participants were entered into the “healthy population” but were, in actuality, far from healthy.
- SAE numbers were left blank.
- Barcodes were missing from samples collected from trial participants.
- The second vaccine dose was administered outside the three-week protocol window.
- New health problems were dismissed as “unrelated” to the vaccination.
- A remarkable number of patients with an observation period of exactly the same duration — 30 minutes, with very little variety in observation times and raising questions as to whether patients were adequately observed or were put at risk.
- Oddities pertaining to the start and end dates of SAEs – for instance, a “healthy” diabetic suffered a “serious” heart attack on October 27, 2020, but the “end” date for this SAE is listed as the very next day, even though the patient was diagnosed with pneumonia that same day.
- Impossible dating: in the aforementioned example of the patient who sustained a heart attack and pneumonia, the individual in question later died, but the date of death is indicated as the day before the patient was recorded as having gone to a “COVID ill” visit.
- Unblinded teams, who were aware of which patients received the actual vaccine or a placebo, were responsible for reviewing adverse event reports, potentially leading to pressure to downplay COVID-related events in the vaccinated, or to indicate that adverse events were related to the vaccine.
- Other adverse events were indicated as “not serious” despite extensive hospital stays, of up to at least 26 days in the case of one patient who suffered a fall which was classified as “not serious,” yet facial lacerations sustained as a result of the fall were attributed to hypotension (low blood pressure).
Many of these practices seem to appear in the trial-related documents released this month.
Medical and scientific experts who spoke to The Defender expressed similar concerns about what this month’s tranche of documents reveals, and addressed cases of “disappearing” adverse events.
Brian Hooker, chief scientific officer for Children’s Health Defense, remarked:
“I’m most concerned about ‘disappearing’ patients. One cannot conduct a valid trial and simply omit the results that they don’t like!
“With the stories about Maddie de Garay and Augusto Roux surfacing, I have to wonder how many other participants were dropped in order to hide vaccine adverse events/effects.
“If you look at the data in VAERS [Vaccine Adverse Event Reporting System], COVID-19 vaccines are the most dangerous ever introduced into the population.”
Dr. Madhava Setty, a board-certified anesthesiologist and senior science editor for The Defender, said:
“The ‘unrelated’ label the investigators use to divert attention from AEs [adverse events] is a powerful point that stands on its own. We haven’t pushed back on this enough.
“Equivalently, we can say that the meager and short-lived benefit of these shots is also ‘unrelated’ using their ‘standards.’ On what grounds can they say that their product is preventing infection (which it isn’t anymore), or death (marginally)?
“They cannot have it both ways. They cannot claim a benefit through short-term outcomes while denying that side effects of any kind are related to their product.
“That’s the whole point of doing a trial. You cannot prove causation, only statistically significant correlation.”
Setty provided further context for his remarks in an April 2022 article for The Defender and in a March 2022 presentation, in which he discussed the number of these adverse events and how Pfizer swept them away (timestamp 24:00).
In Setty‘s view:
“There’s a high likelihood of malfeasance going on. [Pfizer whistleblower] Brook Jackson says the PIs [principal investigators] were unblinded. If true, it would make it very easy for the investigators to bump up the AEs in the placebo group while ignoring some of the AEs in the vaccine group.
“Pfizer claims that 0.5% of placebo recipients suffered a serious adverse event compared to 0.6% in the vaccine group. This is how these events were obscured.”
The extant body of evidence indicates Pfizer “is hiding critical information from regulators,” Setty said:
“The clincher is in the memorandum to the VRBPAC [Vaccines and Related Biological Products Advisory Committee] (Table 2, efficacy populations), where they show us that five times more people in the vaccine group were pulled out of the trial than the placebo within seven days of their second shot for ‘important protocol deviations.’
“In a trial that big the chances that could have happened coincidentally is infinitesimally small (less than 1 in 100,000).
“Moreover, months later, the same thing happened in the pediatric trial (Table 12). This time, six times more children were pulled from the trial after their second dose.
“There are, of course, procedural differences when administering a placebo versus the mRNA vaccine, but why didn’t it happen after the first dose as well?
“Mathematically, that is about as close as you can get to eliminating any ‘shadow of doubt.’ With a formal allegation by a trial coordinator that states the same thing [referring to whistleblower Brook Jackson], we can be assured Pfizer is hiding critical information from regulators.”
BioNTech trials in Germany claim few adverse events ‘related’ to vaccine
The BioNTech trial in Germany tested various dosages of two COVID-19 vaccine formulas, labeled BNT162b1 and BNT162b2 — the latter granted EUA by the FDA.
The latest cache of Pfizer documents suggests a pattern, similar to the one in the U.S. trials, of not reporting adverse events as related to the vaccine.
According to the third interim report, dated March 20, 2021, among trial participants who were administered the BNT162b2 candidate vaccine granted EUA in the U.S.:
- 87% of younger participants reported solicited local reactions, and 88% reported solicited systemic reactions, with 10% reporting solicited systemic reactions of Grade 3 or higher.
- 87% of younger participants experienced “mild” solicited local reactions, and 35% experienced “moderate” solicited local reactions.
- 88% of younger participants experienced “mild” solicited systemic reactions, and 38% experienced “moderate” solicited systemic reactions. As stated in the report:
“The most frequently reported solicited systemic reactions of any severity were fatigue (n=40, 67%), followed by headache (n=32, 53%), malaise (n=24, 40%), and myalgia (n=23, 38%). The remaining symptom terms were less frequent.
“For nausea, headache, fatigue, myalgia, chills, arthralgia and malaise each symptom was assessed as severe in <10% of participants.”
- 43% of younger participants reported a total of 51 unsolicited TEAEs (treatment-emergent adverse events, referring to conditions not present prior to treatment or that worsened in intensity after treatment) within 28 days of the first or second dose, nine of which were deemed to be “related” to the vaccination. One participant in this category sustained a TEAE assessed as Grade 3 or higher, but “which was assessed as not related by the investigator.”
- TEAEs among younger participants included hypoaesthesia, lymphadenopathy, heart palpitations, external ear inflammation, blepharitis, toothache, non-cardiac chest pain, cestode infection, oral herpes, tonsillitis, neck pain, insomnia, anosmia and dysmenorrhea.
- No unsolicited treatment-emergent serious adverse events (TESAEs) or deaths were reported among younger participants, but one discontinued participation due to moderate nasopharyngitis.
- One younger participant “discontinued due to a moderate AE (nasopharyngitis).”
- 86% of older participants reported solicited local reactions, with 6% reporting solicited local reactions of Grade 3 or higher, 78% reporting “mild” solicited local reactions and 36% reporting “moderate” solicited local reactions.
- 72% of older participants reported solicited systemic reactions, with 11% of these participants sustaining solicited systemic reactions of Grade 3 or higher, 69% sustaining “mild” solicited reactions and 36% sustaining “moderate” solicited reactions.
- 33% of older participants reported a total of 20 unsolicited TEAEs, four of which were determined to be “related” to the vaccination. Among older participants, 8% reported a TESAE of Grade 3 or higher, with “one event assessed as related by the investigator.”
- One older participant was reported to have sustained a “not related TESAE” (an ankle fracture).
- TESAEs among older participants included back pain, chest pain, facial injury, increased lipase, increased amylase, muscle spasms, musculoskeletal pain, tendon pain, orthostatic intolerance, renal colic, seborrhoeic dermatitis and “painful respiration.”
Among trial participants who received the BNT162b1 candidate vaccine (not granted EUA):
- 86% of “younger participants” reported solicited (expected) localized reactions (remaining in one part of the body), with 18% reporting Grade 3 or higher solicited local reactions, 86% of younger participants reporting “mild” solicited local reactions and 54% reporting “moderate” solicited local reactions.
- 92% of younger participants reported solicited systemic reactions (spreading to other parts of the body), with 44% reporting Grade 3 or higher solicited systemic reactions, 90% reporting “mild” solicited systemic reactions and 74% experiencing “moderate” solicited systemic reactions.
The report states:
“The most frequently reported solicited systemic reactions of any severity were fatigue (n=68, 81%), headache (n=66, 79%), myalgia (n=51, 61%), malaise (n=50, 60%), and chills (n=47, 56%). The remaining symptom terms were less frequent.
“For nausea, vomiting, diarrhea, myalgia, arthralgia and fever each symptom was assessed as severe in ≤10% of participants.”
- 45% of younger participants reported a total of 83 unsolicited (unexpected) TEAEs within 28 days of receiving the first or second dose.
A total of 51 of these unsolicited TEAEs were reported as “related” to the vaccination, while 2% of participants sustained Grade 3 or higher TEAEs (four in total), “of which three events were assessed as related by the investigator.”
No unsolicited TESAEs or deaths were reported in this category.
- According to the report, among younger participants, TEAEs included:
“‘General disorders and administration site conditions’ reported by 9 participants (11%),” including influenza-like illness and injection site hematoma.
“‘Nervous system disorders’ reported by 10 participants (12%),” including presyncope, hyperaesthesia, paraesthesia, and headache.
“‘Respiratory, thoracic and mediastinal disorders’ reported by 9 participants (11%),” including cough and oropharyngeal pain.”
Other symptoms included back pain, musculoskeletal chest pain, cervicobrachial syndrome, taste disorder, sleep disorder, depression, hallucination, dysmenorrhoea, pruritus and pityriasis rosea, while one participant required the excision (removal) of a papilloma.
- One younger participant discontinued participation in the trial, “due to a moderate AE (malaise),” while another participant discontinued participation “due to dose-limiting toxicity.”
- 83% of “older participants” reported solicited local reactions, but none were reported as Grade 3 or higher, while 83% of solicited local reactions were “mild” and 42% were “moderate.”
- 92% of older participants reported solicited systemic reactions, with 28% of participants experiencing Grade 3 or higher solicited systemic reactions, 89% experiencing “mild” solicited systemic reactions, and 61% experiencing “moderate” solicited systemic reactions.
According to the report:
“The most frequently reported solicited systemic reactions of any severity were headache (n=29, 81%), fatigue (n=27, 75%), myalgia (n=18, 50%), and malaise (n=18, 50%). The remaining symptom terms were less frequent.”
- 36% of participants reported a total of 24 unsolicited TEAEs within 28 days of the first or second dose, nine of which were assessed as “related” to the vaccination.
Of the participants in this category, 11% reported TEAEs of Grade 3 or higher (four events in total), with one of these events assessed as “related” to the vaccination.
- TEAEs reported by older participants included oropharyngeal pain, nasopharyngitis, bladder dysfunction, sleep disorder, musculoskeletal pain and musculoskeletal chest pain, pollakiuria, migraine, syncope and alopecia.
- One older participant receiving the BNT162b1 candidate sustained a TESAE (syncope), and there were no deaths in this category.
Of note, none of the participants for either vaccine candidate were pregnant, which raises questions about recommending and administering the vaccine to pregnant women despite the absence of any clinical trial data.
As the documents show, a wide range of adverse effects were reported, including cardiovascular and nervous system conditions, most of which were determined to be unrelated to the vaccination itself.
Michael Nevradakis, Ph.D., is an independent journalist and researcher based in Athens, Greece.
© 2022 Children’s Health Defense, Inc. This work is reproduced and distributed with the permission of Children’s Health Defense, Inc. Want to learn more from Children’s Health Defense? Sign up for free news and updates from Robert F. Kennedy, Jr. and the Children’s Health Defense. Your donation will help to support us in our efforts.
Featured Video

Iran War Is Accelerating the End of US Dominance
or go to
Aletho News Archives – Video-Images
From the Archives
Gurus of the progressive community . . . Chomsky and Goodman
By Dave Alpert | Intrepid Report | May 23, 2016
There was a time when I, like tens of thousands of my progressive partners, held Noam Chomsky and Amy Goodman in awe. After all, Amy informed us and Noam spoke for us, coherently explaining the issues. However, as I became more aware and more informed, I realized that there were great differences between their thinking and mine.
In many instances, our gurus spoke with forked tongue. Although Amy’s program Democracy Now! was informative, there were many areas of reporting that were out of bounds and were not reported on.
One could legitimately claim that reporters cannot report on everything and they would be right. But let us be honest. When 9/11 occurred, it was an historical event and an event that changed the course of history. Where was Amy? Relatively silent. She invited David Ray Griffin, who has written several books illustrating the lies and misdirections of the government’s narrative about that day, to Democracy Now! which one could claim was a significant journalistic move.
However, instead of interviewing him so that he could reveal to her listening audience the facts that he had accumulated that put into question the government’s explanations of that day, she paired him with a pro-government guest who spent the hour attacking Griffin personally and ignoring any of the data Griffin produced. It became a three-ring circus and helped sabotage any impetus the Truth Movement might have gained within the progressive community. Was that her goal? I’m not sure I can answer that but it was a successful strategy, progressives seemed reluctant to support the Truth Movement. The Movement was being portrayed as one in which there were marginal “conspiracy nuts” leading the charge and should be avoided. … continue
Blog Roll
 Aletho News
Aletho News- Airlines Suffer Losses Estimated at $53Bln Due to Middle East Conflict
- Have you heard the latest joke about Trump and Iran?
- Trump signals possible wind-down of aggression against Iran despite unresolved Hormuz crisis
- The American Fantasy of Iranian Surrender
- US sends more Marines to Middle East as Trump hints at wind-down; contradiction reflects face-saving bid to unsustainable war
- Pentagon Fast Tracks Iran War Ground Option
- Angela Lipps Spent 108 Days in Jail Because a Facial Recognition Algorithm Was Wrong
- Trump White House plagiarized Iran war manifesto from Israel-aligned think tank
- Western media whitewashes Israel’s attempted murder of British journalist in Lebanon
- Israel-linked arms facility set ablaze in EU
- If Americans Knew
- Trump demands trillions in payments from Gulf countries, billions from Harvard – Not a ceasefire Day 162
- Meet the former fashion blogger and shady doctor behind the ‘30,000 dead’ Iran psy-op
- Vatican Secretary of State to Trump, Israel: End the war as soon as possible
- The Majority of Americans Believe War Against Iran Benefits Israel More Than US
- Efforts to shut down pro-Palestinian speech face series of setbacks in court
- What has Israel done to my brother?
- Eid without worship in Al Aqsa – Not a ceasefire Day 161
- Trump: Israel attacked Iranian gas field without US knowledge. No more such attacks!
- America’s friends must help extricate it from an unlawful war
- Democratic Nat’l Committee’s Kneejerk Backing of Israel Is Political and Moral Failure
- No Tricks Zone
- Energy Expert: Germany’s Nuclear Phaseout Was A “500 Billion Euro Mistake”
- New Research: South Australia’s Mid-Holocene Sea Surface Temperatures Were 4°C Warmer Than Today
- Storing Green Energy To Last Germany 10 Days Would Require A 60-Million Tonne Battery
- New Studies: UK Sea Levels Were 4 Meters Higher Than Today During The Mid-Holocene
- Destructive Green New Deal: German Energy And Metal Group Warns Of Drastic Crisis
- New Study Documents A 20-Year Pause In Arctic Sea Ice Decline – Driven By Internal Variability
- Wake-up Call: Survey Shows Majority Of Germans Now Favor Postponing Climate Targets!
- Televised! Leading German Political Candidate Tells Schoolchildren CO2 Makes Sun Hotter!
- New Study: A Century Warming Of 1.1°C Is ‘Commonplace’ And ‘Not Unusual’ During This Interglacial
- New Study: ‘Internal Noise’ And Volcanic Forcing Can Trigger 10-15°C Warming Within Decades
