CHD Scientist: CDC, FDA COVID Vaccine Safety Monitoring ‘Insulting, and Many People Are Injured’
By Suzanne Burdick, Ph.D. | The Defender | April 29, 2026
Federal health officials under the Biden administration failed abysmally to look for COVID-19 vaccine safety signals, according to congressional testimony delivered today by Children’s Health Defense (CHD) Senior Research Scientist Karl Jablonowski.
The government’s vaccine safety monitoring “over the past several years has been insulting, and many people are injured,” Jablonowski wrote in his written testimony.
History repeats itself if we don’t learn our lessons, Jablonowski warned.
“The COVID-19 pandemic created over 100 billionaires in the United States and over 1,000 billionaires around the world,” Jablonowski wrote. “Anything that profitable is going to repeat.”
Jablonowski, who holds a doctorate in biomedical and health informatics from the University of Washington’s School of Medicine, spoke as a witness at the U.S. Senate Permanent Subcommittee on Investigations hearing, “Unmasked: How Biden Health Officials Purposely Turned a Blind Eye Toward COVID-19 Vaccine Safety Signals.”
Hours before the hearing, Sen. Ron Johnson (R-Wis.), subcommittee chair, released a report detailing how Biden-era federal health officials refused to use a state-of-the-art statistical tool for detecting COVID-19 vaccination signals in VAERS — even though they knew the tool they were using was too broken to pick up on safety signals, including sudden cardiac death.
Johnson’s report, which cited roughly 600 pages of emails, revealed that in 2021, officials with the U.S. Food and Drug Administration (FDA) told an FDA researcher to “cease and desist” using the state-of-the-art tool to analyze COVID-19 vaccine injury reports in the Vaccine Adverse Event Reporting System (VAERS).
Johnson obtained the emails after he subpoenaed the U.S. Department of Health and Human Services in January 2025 for COVID-19 vaccine safety records and pandemic-related communications.
FDA was ‘blind’ to COVID vaccine injury reports in VAERS
In his testimony, Jablonowski detailed how each of the federal government’s three vaccine safety monitoring systems — VAERS, V-safe and Vaccine Safety Datalink (VSD) — had “pitfalls” and “failed” to adequately assess safety issues with the COVID-19 vaccine and other vaccines.
The failures of vaccine safety monitoring “can be, and were, catastrophic,” he said.
For instance, the FDA insisted on monitoring COVID-19 vaccine reports using a method that it knew didn’t work. The FDA knew the method was likely to give inaccurate results if similar vaccines — such as the Pfizer and Moderna COVID-19 vaccines — were included in the dataset. This is called masking.
“The FDA was completely blind to COVID-19 vaccine adverse events,” Jablonowski wrote. He said the FDA could have used an improved statistical method accounting for masking.
A 2022 peer-reviewed paper in Drug Safety showed that the improved method detected roughly 25 statistically significant COVID-19 vaccine safety signals — including sudden cardiac death, Bell’s palsy and pulmonary infarction — that the FDA’s older method missed.
In an earlier interview with The Defender, Jablonowski explained why it was so harmful for the FDA to continue using the older method:
“Imagine a night watchman has to find something on the ground. But instead of holding a flashlight, he is wearing sunglasses. In the morning, he says he didn’t find anything. That’s true, but it’s because he was using a tool that impeded his ability to see.”
As of March 27, 1,675,590 adverse events were reported to VAERS following COVID-19 vaccination, according to OpenVAERS. That number includes over 39,077 reports of death, 29,200 reports of myocarditis or pericarditis, and 18,009 reports of Bell’s palsy.
A national survey conducted in November 2025 found that roughly 1 in 10 U.S. adults who received the COVID-19 vaccine experienced “major” side effects.
V-safe was designed to collect ‘inconsequential’ data
Jablonowski told lawmakers that the Centers for Disease Control and Prevention’s (CDC) COVID-19 vaccine safety monitoring tool, V-safe, was designed to collect only “inconsequential” information that no one really cares about.
The V-safe app invited COVID-19 vaccine recipients to check off boxes to indicate what, if any, side effects they experienced after getting the shot.
However, the box options were for common short-term vaccine side effects that most people would consider “inconsequential,” such as chills, headache, joint pain, muscle or body aches, fatigue or tiredness, nausea, vomiting, diarrhea, abdominal pain or rash.
If a person experienced a more serious problem, they had to manually type it into the “other” text field, Jablonowski noted. He said:
“It is with horror that we find 366 individuals typed ‘myocarditis’ in the ‘other’ free-text field, a condition requiring a medical diagnosis. The horror is amplified by the nearly 50,000 registrants who typed ‘chest pain’ into the ‘other’ free-text field.”
Vaccine Safety Datalink is off-limits to independent researchers
Jablonowski also detailed how VSD, a collaborative database of patient information from 13 integrated healthcare organizations covering over 15.5 million people, also fails the public.
VSD data can ostensibly be used to detect vaccine safety issues in near-real time, Jablonowski said.
The problem is that only a small handful of scientists are ever allowed to look at the data. Jablonowski said:
“This many million-dollar taxpayer funded resource is not available to any scientist outside of the 13 Managed Care Organizations (MCO) or the federal government without independent IRB [independent review board] applications approved by all 13 MCOs, an estimated $250,000 per project.”
In other words, independent researchers are realistically barred from analyzing the data. “Transparency is simply unattainable,” Jablonowski said.
Watch Jablonowski’s opening statement here.
This article was originally published by The Defender — Children’s Health Defense’s News & Views Website under Creative Commons license CC BY-NC-ND 4.0. Please consider subscribing to The Defender or donating to Children’s Health Defense.
FDA Won’t ‘Rubber-Stamp’ Pfizer mRNA Flu Vaccine Without Better Safety Data
By Michael Nevradakis, Ph.D. | The Defender | December 15, 2025
The U.S. Food and Drug Administration (FDA) likely won’t approve Pfizer’s mRNA flu vaccine unless the drugmaker produces data proving the product is safe for seniors, according to FDA Commissioner Marty Makary.
Makary told Fox News last month that the data from Pfizer’s recently completed Phase 3 clinical trial showed that adults 65 and older were at higher risk of several serious adverse events, including kidney failure and acute respiratory failure.
“We’re not just going to rubber-stamp new products that don’t work, that fail in a clinical trial,” Makary said. “It makes a mockery of science if we’re just going to rubber-stamp things with no data.”
Makary said the shot “failed in seniors” and the trial data “showed zero benefit” from the vaccine.
Karl Jablonowski, Ph.D., senior research scientist for Children’s Health Defense, said Makary’s comments signal a change in the way the FDA evaluates clinical trial data for vaccines.
“Makary’s FDA threw out the rubber stamp,” Jablonowski said. “The FDA, under different leadership, may have brushed off the lack of efficacy and Pfizer’s concerning safety data. A future administration may resurrect the rubber stamp. For the time being, this is Makary’s FDA.”
Last month, Pfizer published the results of its clinical trial in the New England Journal of Medicine (NEJM). However, the published results included data only for participants between 18 and 64. Data for participants 65 and older, published only on ClinicalTrials.gov, drew criticism from some scientists.
That data showed that elderly trial participants who received the mRNA vaccine had a significantly higher rate of death and several serious adverse events, including cancer, compared to participants who received the conventional non-mRNA flu shot.
This contrasts with Pfizer’s claims that the vaccine delivered “statistically superior efficacy” compared to the conventional flu shot, and that the frequency of serious adverse events was “similar” across the mRNA and non-mRNA groups.
“The disposition of the kidney and lung issues associated with the mRNA shot was concerning,” Jablonowski said.
Some experts noted that even among the 18-64 age group, adverse events were higher among trial participants who received the mRNA shot.
The only mention of the trial data for people 65 and over in the NEJM came in an accompanying editorial, which noted that this age group faces “the highest risk of hospitalization or death” from the flu.
Dr. Meryl Nass, a former internist and founder of Door to Freedom, said she was encouraged by Makary’s remarks. She said the FDA is legally required to license only those drugs that are proven to be safe and effective.
“This mandate is at least 70 years old,” Nass said. “What Makary is saying is already mandated by Congress. But the FDA has chosen to ignore that mandate due to politics, and Congress has failed to enforce it. Makary is actually obeying the law for the first time in decades regarding flu shots.”
Makary: annual mRNA vaccination ‘not based on science’
Makary told Fox News that past administrations rubber-stamped vaccine approvals even when safety data was questionable.
“That was the MO in the Biden administration with the eternal COVID booster approvals for young healthy kids,” Makary said.
The current administration will adopt a different approach to vaccine approvals, especially for children, Makary said.
“Recommending that a 6-year-old girl get another 70 mRNA COVID shots, one each year for the rest of her life, is not based on science,” Makary said.
Makary’s remarks came days after the release of a leaked memo in which Dr. Vinay Prasad, director of the FDA’s Center for Biologics Evaluation and Research, said changes are coming to the framework for evaluating flu vaccines.
“We will revise the annual flu vaccine framework, which is an evidence-based catastrophe of low quality evidence, poor surrogate assays, and uncertain vaccine effectiveness measured in case-control studies with poor methods. We will re-appraise safety and be honest in vaccine labels,” Prasad wrote in that memo.
Dr. Robert W. Malone, a member of ACIP and the committee’s influenza workgroup, told The Epoch Times that Prasad’s memo means “the entire influenza vaccine, annual vaccination enterprise is now subject to major disruption.”
In May, COVID-19 vaccine manufacturer Moderna withdrew its application for FDA approval of a combination mRNA flu and COVID-19 vaccine, after the FDA requested more clinical trials.
In June, the CDC’s vaccine advisers voted to stop recommending flu shots that contain thimerosal — a mercury-based preservative linked to neurodevelopmental disorders, including autism.
‘No one has figured out’ how to make mRNA shots safe
Makary’s statements came amid growing questions about the safety, efficacy and necessity of existing non-mRNA flu vaccines and waning uptake of the shots.
A Cleveland Clinic study published in April found that people who received the flu vaccine were 27% more likely to get the flu than those who didn’t.
Another study, published that month in JAMA Network Open, found that flu vaccines, whether given alone or in conjunction with COVID-19 shots, caused women to have longer menstrual cycles.
Endpoints News reported last month that public demand for flu vaccines is stalling and that “the general consensus among vaccine makers for Covid-19, flu and RSV is that dampening demand has shrunk sales.” Data from Eurostat indicate a decline in flu vaccine uptake in the European Union.
Research into mRNA-related platforms is also facing growing scrutiny. In August, the U.S. Department of Health and Human Services cancelled nearly $500 million in funding for mRNA vaccine research.
“With regard to mRNA injections, no one has figured out how to make them safe,” Nass said. “mRNA shots provide an unknown dose, and they can be ‘the gift that keeps on giving,’ because we don’t know how to shut off the production of mRNA-coded proteins. We probably never will.”
Nass added that while FDA rules require that a specific dose be established for every drug, “somehow this rule has never applied to mRNA vaccines.”
“I believe the mRNA platform is irrevocably flawed for this reason alone, although there are other toxicities involved that also make the platform problematic,” Nass said.
A growing number of scientists have called for the suspension or withdrawal of the administration of mRNA vaccines and products.
This article was originally published by The Defender — Children’s Health Defense’s News & Views Website under Creative Commons license CC BY-NC-ND 4.0. Please consider subscribing to The Defender or donating to Children’s Health Defense.
Vaccine injury lawyer delivers scathing rebuke of childhood vaccine schedule — Offit, Hotez decline to debate
By Michael Nevradakis, Ph.D. | The Defender | December 5, 2025
The Centers for Disease Control and Prevention’s (CDC) vaccine advisory committee today heard from a vaccine injury lawyer who proposed the committee revisit the childhood vaccine schedule.
Attorney Aaron Siri told members of the Advisory Committee on Immunization Practices (ACIP) that the vaccines were recommended without sufficient data and that the expansion of the schedule coincided with a rise in chronic illness among U.S. children.
Siri, a vaccine critic and author of “Vaccines, Amen: The Religion of Vaccines,” called for a reexamination of the childhood immunization schedule based on “robust” safety data.
Siri challenged claims that the childhood vaccination schedule has been tested in its entirety, that the vaccines are safe and that routine childhood vaccines have been proven to prevent transmission.
He also questioned claims that scientists have conducted the testing necessary to assert definitively that there is no possible link between vaccines and autism.
Siri recommended ACIP revisit childhood vaccine recommendations based on “robust” clinical trial and post-licensure safety data and called on the committee to respect the “right of informed consent.”
“Mandates make vaccines political” and also “impact those who most need to avoid” certain vaccines, he said. When people report vaccine injuries, members of the medical community “pretend that they don’t exist.”
Siri represents plaintiffs in vaccine-related lawsuits against federal agencies and pharmaceutical companies.
Children’s Health Defense CEO Mary Holland said Siri “accurately compared the pre-1986 Act vaccine schedule with the post-1986 schedule, when doctors and vaccine manufacturers have been absolved from all real responsibility for the vast vaccine injuries they have caused,” Holland said.
The National Childhood Vaccine Injury Act of 1986 granted vaccine makers immunity from liability for most injuries caused by their products.
ACIP didn’t vote on any aspect of the childhood vaccine schedule today. In June, ACIP formed a committee to study the cumulative effect of all vaccines given during childhood.
Key vaccine advocates declined ACIP’s invitation to deliver presentations
Siri’s presentation came shortly after ACIP voted to end the recommendation that all infants born in the U.S. receive the hepatitis B (Hep B) vaccine within 12-24 hours of birth.
The committee also voted to recommend that families determine whether to give their child the Hep B shot at birth through individual decision-making and consultation with their physician.
Siri cited the licensing of Hep B vaccines as an example of flawed studies leading to the licensing of a vaccine. He called those studies “underpowered” and “industry-funded.”
Siri’s presentation stirred controversy even before it began. In a post on X yesterday, Sen. Bill Cassidy (R-La.), chairman of the U.S. Senate Health, Education, Labor & Pensions (HELP) Committee, dismissed Siri’s qualifications and said ACIP “is totally discredited.”
Siri responded that Cassidy’s post was “deeply ironic,” given that vaccine manufacturers are legally protected from lawsuits.
“Childhood vaccines are the only product in America where you cannot ever sue the company that killed or injured your child on the basis the company could’ve made the product safer. If vaccines are so safe, why do they need this protection?” Siri wrote on X.
ACIP member Dr. Cody Meissner called Siri’s presentation a “terrible distortion of all the facts” and said Siri shouldn’t have been invited. Earlier, Meissner voted against the proposal to end the universal Hep B vaccine recommendation for newborns.
ACIP also addressed controversy over Siri’s presentation and the lack of a pro-vaccine counterweight. Mina Zadeh, Ph.D., ACIP’s executive secretary, said the committee “invited several people to give us a broad perspective” on the childhood vaccination schedule.
Those invitees included two prominent and outspoken promoters of vaccines — Dr. Paul Offit and Peter Hotez, M.D., Ph.D. Both declined. Hotez told STAT that Siri “shouldn’t be there in the first place.”
Siri responded that the U.S. has “the worst health outcomes of all developed countries.”
Liability shield disincentivizes vaccine makers from performing proper safety testing
Siri used the opportunity today to criticize the National Childhood Vaccine Injury Act of 1986. He said the liability shield provided by that law disincentivized vaccine manufacturers from focusing on the safety of their products.
“Companies, including pharmaceutical companies, are driven by economic self-interest,” Siri said. “With drugs and non-routine vaccines, they … remain liable for the injuries caused by those products after they come to market and hence, they have an economic self-interest in doing robust clinical trials beforehand.”
“When it comes to routine childhood vaccines … they don’t have those same concerns,” Siri said.
He said the number of vaccines on the childhood schedule skyrocketed — from three to 72 — after Congress passed the 1986 act. Those initial three vaccines “were causing so much harm, all the manufacturers stopped making them or went out of business,” prompting the passage of the act.
“For every other product I’m aware of, the solution is to make a better, safer product. But when it came to these vaccines, Congress went a different way” by giving these companies “unprecedented broad immunity,” Siri said.
‘You can’t find what you’re not studying’
Siri also criticized the shortened clinical trial process for childhood vaccines, which results in recommendations being made on the basis of insufficient data and the inability to detect any long-term health impacts from the vaccines.
“Most recommendations for routine use by ACIP of a particular vaccine happened very shortly after its licensure, and hence the primary data often available for a specific vaccine would have been its clinical trial data,” Siri said.
He also criticized the lack of post-licensure safety monitoring.
“You can’t find what you’re not studying,” Siri said. “When you give a product to a baby or an infant in particular, you often won’t know what neurological, immunological or developmental issues that product can cause until you’ve tracked that child for at least a few years.”
Citing autism as the “injury claimed to be the most thoroughly studied,” Siri said the medical community has not conducted studies that would definitively eliminate a vaccine-autism link, even though the 1986 act listed autism as one of 11 conditions that warrant further study to determine a possible link with vaccination.
“It was a commonly claimed enough injury back in 1986 … to make it on this list of 11 conditions,” Siri said.
U.S. ‘an international outlier’ on childhood vaccination
Today’s meeting also included a presentation by Tracy Beth Høeg, M.D., Ph.D., who earlier this week was named the next leader of the FDA’s Center for Drug Evaluation and Research.
Høeg compared U.S. childhood vaccine requirements and health outcomes with those of her native Denmark. There are “eye-opening differences in the recommendations” between the two countries, she said.
While the U.S. requires 72 core childhood vaccine doses, Denmark requires 11 — in line with most other high-income countries. Høeg said this makes the U.S. “an international outlier” on childhood vaccination.
The higher vaccine load “results in an increased exposure to aluminum,” Høeg said, with U.S. children exposed to 5.9 milligrams (mg) of aluminum by age 2 and 8.0 mg by age 18. In Denmark, the corresponding figures are 1.4 and 2.9 mg, similar to other high-income countries.
While there isn’t “robust enough” data indicating “specific health concerns” resulting from this level of aluminum exposure, Høeg said there also is insufficient data to establish a safe level of exposure.
“We need to admit that we may not know what the side effects of doing this, especially all at once, could be,” she said.
Increased vaccination also hasn’t delivered better health outcomes for U.S. children, according to Høeg. She cited the examples of the Hep B and meningococcal vaccines, which Denmark does not recommend for children, unlike the U.S. Yet, levels of hepatitis B and meningitis among children in the two countries are similar.
Høeg said U.S. health agencies should “avoid overmedicalizing childhood” and owe American children recommendations that are “based on data and not politics.”
Potential risks of post-vaccine aluminum accumulation ‘a warranted concern’
Dr. Evelyn Griffin, an OB/GYN and member of three ACIP work groups called for more research into the safety of aluminum-based adjuvants used in vaccines.
Griffin said aluminum salts are the most widely used adjuvant. Yet, the mechanisms underlying the use of aluminum salts in vaccines “are not fully understood.” She said only one peer-reviewed study has examined the effects of aluminum in infants’ blood following vaccination — but that study used a small sample and didn’t collect long-term data.
According to Griffin, current FDA aluminum exposure limits are increasingly questioned, as “appropriate testing was not performed.” She said recent studies have suggested that aluminum accumulation is “a warranted concern” and called for studies on the long-term impact of aluminum exposure and who is most at risk.
Griffin called on ACIP to determine how it can assess the safety and effectiveness of adjuvants in currently recommended vaccines for all ages, including studies regarding whether multiple aluminum-containing vaccines should be administered on the same day during early infancy.
In October, ACIP announced the creation of a new work group that will study the safety of aluminum adjuvants. ACIP did not hold a vote relating to the aluminum content of vaccines at today’s meeting.
Watch the ACIP meeting here.
This article was originally published by The Defender — Children’s Health Defense’s News & Views Website under Creative Commons license CC BY-NC-ND 4.0. Please consider subscribing to The Defender or donating to Children’s Health Defense.
Whistleblower Biologist Says Pfizer Covered Up Her Exposure to Engineered Virus, Threatened Family
By Brenda Baletti, Ph.D. | The Defender | November 7, 2025
Molecular biologist Becky McClain began raising safety concerns in 2000, soon after she started working in Pfizer’s Biosafety Level 2 lab in Connecticut.
Three years later, after management failed to address the issues, McClain was exposed to a genetically modified lentivirus, engineered using gain-of-function technologies that made the virus more infectious and more pathogenic.
The exposure left her disabled, with symptoms including numbness, periodic paralysis, pain and other neurological problems. Doctors couldn’t diagnose or effectively treat her condition because Pfizer refused to disclose what she had been exposed to, citing “trade secrets.”
The incident launched McClain into a decade-long fight to understand her illness and obtain her exposure records so she could seek proper treatment. During her battle, she became a whistleblower, standing up to Pfizer’s threats against her and her family.
In her new book from Skyhorse Publishing, “Exposed: A Pfizer Scientist Battles Corruption, Lies, and Betrayal, and Becomes a Biohazard Whistleblower,” McClain recounts how she raised workplace safety concerns, suffered exposure to a dangerous virus, fought Pfizer for years in court, and resisted the company’s repeated attempts to silence her — ultimately winning a legal victory.
McClain refused to sign a gag order — even after Pfizer fired her, harassed her and threatened her — making her one of the few people who can share her story publicly.
In her book, McClain exposes corruption she says runs not just through Pfizer, but across the pharmaceutical industry and the agencies meant to hold it accountable — from the Occupational Safety and Health Administration (OSHA) and U.S. Food and Drug Administration (FDA) to the federal courts.
Consumer safety advocate Ralph Nader wrote in his foreword to the book:
“No general description of this book can convey the horror and details of what Becky McClain and her husband, Mark, endured at the hands of Pfizer, enabled over the years by collusion with government officials. Pre-verdict and post-verdict, this company employed thuggish retaliatory tactics, blacklisting, threats, harassments, wrongful discharges, coverups, and demands for total gag orders.
“Those tactics were designed to keep her case from flaring into a national demand for Congressional regulation in the form of rigorous biolab inspections and mandatory safety/health standards with teeth. Against this objective, Pfizer and the bioengineering industry are succeeding.”
‘If you document biosafety issues and or speak out about them, you’re out’
In an interview with The Defender, McClain said she noticed safety issues as soon as she started working in the lab.
“We had no break room, no safe break room. We had unsafe offices. We had improper biocontainment protocols using infectious agents,” she said. “And although the lab was unsafe, management made it worse by instilling a culture of fear for anyone who dared to raise safety issues.”
McClain said most scientists at the lab shared her concerns, but managers made it clear: “If you document biosafety issues and or speak out about them, you’re out.”
Scientists at the lab worked on genomic-altering biotechnologies, creating viruses capable of entering cells and changing their genomes, she said.
After multiple safety incidents — including one that left several scientists sick — McClain walked in one morning to find “a mess” on her personal workbench. A supervisor and an untrained scientist had left a dangerous experiment there overnight, without McClain’s knowledge.
A month later, the untrained scientist asked McClain if she knew anything about lentiviruses, a family of viruses that includes HIV and FIV (feline immunodeficiency virus).
By then, McClain was experiencing numbness on one side of her face, which a neurologist suggested might be the start of multiple sclerosis.
McClain realized she had likely been exposed to a modified lentivirus and asked the scientist to find out more about its safety. He returned “a little bit nervous” and told her the virus he had used on her bench was safe, indicating it wasn’t infectious to humans.
That conversation marked the beginning of McClain’s fight to obtain her exposure records. Pfizer refused to provide them, telling her that “trade secrets supersede your right to that information.”
As her condition worsened, McClain went on medical leave — and the company terminated her.
McClain was shocked because she had assumed worker rights would protect her. She said:
“I couldn’t get directed medical care for my illness, which was a mystery illness because these genetically engineered virus technologies were designed to cause new emerging diseases for use in laboratory research studies.
“So when I visited doctors, no one knew what was happening. They were all fearful and unable to explain my illness.
“My husband and I feared I was going to die. It eventually became very, very, very, very severe. It began with numbness on the left side of my face, then extreme left jaw pain, inflammation of my trigeminal nerve, headaches, spinal pain, then periodic paralysis.”
‘There’s no free speech for scientists’
McClain turned to OSHA for help, submitting documentation she had gathered that exposed egregious safety violations in the lab. OSHA refused to help her access her exposure records and didn’t even conduct a safety inspection of the lab.
“OSHA is a captured agency now,” McClain said. “They oversee approximately 24 different whistleblower laws under one roof, making it easy for the industry to control OSHA. It’s easy to capture. Place a corporate head to oversee OSHA, and you gain control of all the whistleblower laws and investigations.”
After OSHA declined to provide substantive help, McClain’s next step was clear. “The only legal remedy to get my exposure records was to file a civil whistleblower claim,” she said.
During the process, McClain met countless other scientists in similar situations.
“There’s no free speech for scientists,” she said. She cited examples of scientists being censored and smeared as “anti-vaxxers” during the COVID-19 pandemic, when “they were merely raising legitimate safety concerns.”
A recent investigation by The Defender found that OSHA told healthcare employers not to report employees adverse reactions to COVID-19 vaccines — but to continue reporting injuries caused by all other vaccines.
Pfizer launched ‘backdoor retaliation’ by targeting McClain’s husband
Throughout her long legal battle, Pfizer tried relentlessly to compel her to sign a gag order. She refused, knowing that signing would cost her the leverage she needed to access information about her exposure.
The company launched what McClain called “backdoor retaliation” by targeting her husband, who worked at the FDA in Connecticut.
“Two months before the trial, my husband was called into his office and told that if he didn’t make me settle with Pfizer, he’d be out of a job,” McClain said.
The threat terrified the couple, as McClain was extremely sick and they relied entirely on his income. “I thought Pfizer couldn’t have that kind of reach … my husband works for the government. But they did,” she said.
Her husband refused to force her to sign a gag order. After facing false accusations despite a spotless 18-year record as a commissioned officer, he left the FDA.
McClain eventually won her free speech whistleblower lawsuit in a 2010 jury trial, even though later revelations showed that the judge had financial conflicts of interest. She received 10 years of back pay — but no compensation for her exposure, illness or suffering.
Pfizer faced no obligation to remediate its safety program.
Although McClain never gained full access to her exposure records, she did obtain additional details about the virus, which she explains in her book.
Today, she publicly advocates for industry reform. She told The Defender there are several key issues she thinks need to be addressed. She said:
“First, is that all gag orders related to lab injuries and public health and safety concerns should be illegal. The public has a right to know about the dangers in these laboratories, especially in our post-pandemic environment.
“Then, OSHA needs to be revamped. It’s a captured agency.”
McClain added that OSHA can’t effectively oversee biotechnology because the agency doesn’t fully understand the serious and unique safety risks. She said the safety problems run through biotechnology research in academia, government and the private sector — each with its own set of regulations — and that the private sector faces the fewest rules.
“The bottom line is that we need better free speech and whistleblower protections for scientists, physicians, and injured workers,” McClain said. “No one should go through 10 years of hell just to have a safe workplace or to protect the public by standing up for professional standards.”
This article was originally published by The Defender — Children’s Health Defense’s News & Views Website under Creative Commons license CC BY-NC-ND 4.0. Please consider subscribing to The Defender or donating to Children’s Health Defense.
Tylenol, FDA Knew About Autism Risk For Years, Newly Surfaced Emails Show
By Brenda Baletti, Ph.D. | The Defender | September 29, 2025
Makers of Tylenol and the U.S. Food and Drug Administration (FDA) knew for years about the likely association between the drug’s use during pregnancy and neurodevelopmental disorders, including autism, according to documents obtained in lawsuits against Kenvue.
“The weight of evidence is starting to feel heavy to me,” Rachel Weinstein, U.S. director of epidemiology for Johnson & Johnson’s (J&J) pharmaceutical division Janssen, said in an email commenting on several studies showing the link.
Daily Caller News Foundation obtained the emails from Keller Postman LLC, the law firm representing plaintiffs in a federal class action lawsuit against Kenvue.
J&J made Tylenol until 2023, when it spun off production to Kenvue, a separate company.
The email revelations follow President Donald Trump’s announcement last week that pregnant women should not take Tylenol, and the FDA’s announcement that it will add warnings to products containing acetaminophen.
The updated product labels will warn that acetaminophen may be associated with a higher risk of neurological conditions, including autism and attention-deficit/hyperactivity disorder (ADHD), in children. The FDA said it will also warn physicians and the public about the risk.
Mainstream media and public health organizations attacked the warnings as unfounded or overblown. Some news organizations quoted scientists — like University of Massachusetts epidemiologist Ann Bauer — who published studies identifying the link between Tylenol and autism and called for warnings, but who are now publicly backpedaling on their concerns.
However, the Daily Caller found that despite confusion in the media and among public health experts, emails show that as early as 2008, officials at J&J were privately concerned about credible evidence of a possible link between autism and acetaminophen. They acknowledged the link in an email and suggested further investigation.
Internal FDA meta-analyses shared with The Defender show that the agency had for years considered adding new warnings about acetaminophen’s side effects for children.
In 2019, FDA scientists conducted a meta-analysis that found urogenital disorders in infants linked to the drug. The scientists also noted links to neurodevelopmental issues. In 2022, the FDA conducted another meta-analysis that found a link to ADHD.
Tylenol makers ‘closely tracked a drumbeat of scientific publications’ showing link to autism
The Daily Caller News Foundation obtained emails spanning more than a decade indicating that company insiders at J&J had been alerted about the possible link between acetaminophen and neurological disorders. The emails showed J&J even considered pursuing further research, but decided against it.
The outlet also obtained a 2012 email by Leslie Shur, head of the division at J&J that monitors side effects, acknowledging another consumer complaint about the issue, and a 2014 email showing that the issue was raised with CEO Alex Gorsky, whose name is misspelled in the email.
According to journalist Emily Kopp, who wrote the Daily Caller story:
“The makers of Tylenol have closely tracked a drumbeat of scientific publications finding an association between taking the blockbuster drug in pregnancy and infancy and autism risk, other company documents show.
“A 2018 internal presentation the company labeled ‘privileged and confidential’ acknowledges that observational studies show a ‘somewhat consistent’ association between prenatal exposure to Tylenol and neurodevelopmental disorders.
“Another presentation slide acknowledges that larger meta-analyses — reviews summarizing multiple scientific studies — found an association, but notes weaknesses of these studies like confounding variables and subjectivity in measuring autistic traits.”
A Kenvue spokesperson told the Daily Caller that the company believes there is “no causal link between acetaminophen use during pregnancy and autism” and that its projects are “safe and effective” when used as directed on the label.
Kopp noted the company’s website also states that “credible, independent scientific data continues to show no proven link between taking acetaminophen and autism,” and that “there is no credible science that shows taking acetaminophen causes autism.”
Yet, she found that internal emails showed employees discussing a 2018 study and a 2016 study that both concluded pregnant women should be cautioned about the possible effects of taking Tylenol while pregnant.
She also found emails indicating that J&J considered funding studies on Tylenol’s possible link to autism, but decided against “sticking their necks out,” worried their studies could confirm the findings.
According to Kopp:
“The company also conducted research it described as ‘social listening’ by tracking Google searches and social media posts seeking evidence about Tylenol and autism from January 2020 through October 2023.
“The company initiated the social media trends research after the 2021 publication of a call to action on Tylenol in Nature Reviews Endocrinology by 13 U.S. and European experts ‘in light of the serious consequences of inaction.’”
The company wrote a 2023 review, Project Cocoon, which reported on concerns with urinogenital and neurological side effects of the drugs in babies, which executives noted touches“every aspect of the brand,” Kopp wrote.
FDA also concerned with mounting evidence
The FDA also grew concerned with the mounting evidence of a link between acetaminophen and neurodevelopmental disorders, beginning with a publication in JAMA Pediatrics in 2014 and followed by several major publications over the next several years, according to psychiatrist David Healy.
Healy is an expert witness in a case against Kenvue and Safeway, alleging they failed to adequately warn consumers about the risk of autism or ADHD from prenatal exposure to the drug.
Documents from 2019 and 2022, made available through Freedom of Information Act requests associated with the lawsuit and shared with The Defender, show that based on meta-analysis of the published literature, the FDA identified consistent links between acetaminophen and both urogenital and neurodevelopmental risks.
As early as 2019, FDA study authors recommended that the labels be revised to advise pregnant women to “be careful about casual use of acetaminophen when it is not strongly needed for pain or other purposes.”
The 2022 document, focused largely on neurological outcomes, states that despite study limitations, meta-analyses and other research consistently found links between acetaminophen and ADHD, and as a result, “it may be prudent, as a precautionary measure …” However, the rest of the recommendation is redacted.
Healy said the revelations by Weinstein and others working with J&J are particularly significant because drugmakers have the responsibility to inform consumers when they know a drug may be linked to an adverse event.
“The onus to warn does not arise when there is a clear cause and effect,” Healy said. “It arises when there are grounds to think there might be a problem.”
This article was originally published by The Defender — Children’s Health Defense’s News & Views Website under Creative Commons license CC BY-NC-ND 4.0. Please consider subscribing to The Defender or donating to Children’s Health Defense.
The Dark History of Hormone and Puberty Blockers
Again and again, these drugs are pushed on defenseless patients because of how much money they make
A Midwestern Doctor | The Forgotten Side of Medicine | September 14, 2025
Story at a Glance:
• Puberty blockers used for transgender medicine belong to a class of drugs (GnRH agonists) which permanently block the production of sex hormones in the body. As hormones are essential for the body, GnRH agonists are amongst the most harmful drugs on the market.
• Originally approved (with grave reservations from the FDA) as a palliative treatment for severe prostate cancer, these drugs (e.g., Lupron) have rapidly proliferated into a wide range of areas in medicine, including routine prostate cancer, a myriad of female issues (e.g., endometriosis) and all sorts of experimental uses on children (e.g., making them taller).
• This proliferation was due to manufacturers pricing the drugs to generate enormous profits for themselves and doctors (in many cases constituting most of urology practices’ revenues)—likely why most urologists, when surveyed, admitted prescribing Lupron despite not believing it worked.
• These drugs rapidly age the body, causing permanent and crippling side effects, including severe bone loss, pain, soft tissue damage, severe pain hormonal disruption, sexual dysfunction, psychiatric issues, cognitive impairment, cancer risks, and cardiovascular and gastrointestinal disorders.
• Their use in transgender children to block puberty stems from an unproven theory that it ultimately leads to a more satisfying gender transition in adulthood. However, while aggressively advocating for them and publicly claiming these drugs are safe, effective, and reversible, in private, the group authoring the medical guidelines have admitted they have no idea what they are doing and know there are serious safety issues with the drugs.
• This article will expose the hidden truths about hormone blockers, the forgotten generations whose lives were ruined by them, and extensive documentation showing how dangerous these ‘safe and effective’ drugs are.
Transgenderism has rapidly become one of the most contentious political issues in our country and due to its rapid rise, a variety of theories have been put forward to explain where it emerged from. Remarkably, I almost never see what I believe to be one of the most important facets of the topic discussed—the immense dangers of hormonal blockers routinely used in this field or the appalling history of these drugs and how again and again, they’ve been thrust into new markets they had no place ever being used in because of how profitable they are.
As such, when laws are periodically passed banning their use in children (which has now happened in many Red States), I rarely see the actual dangers of these drugs discussed, and when I’ve spoken to left-wing colleagues (including pediatricians) opposing these laws about the topic, most are genuinely unaware the drugs have negative side effects. Because of this, I believe it is vital to expose the actual truth behind these drugs.
How Hormonal Blockers Work
There are a variety of ways you can block the production of hormones in the body. Since the signal to produce sex hormones (e.g., estrogen and testosterone) begins in the brain, cutting that signal off mostly eliminates the body’s production of hormones. The most powerful hormonal blockers, the GnRH agonists, work by overstimulating the brain’s GnRH receptors so that they becomes “burned out” and no longer respond to the natural release of GnRH in the body, thereby short-circuiting the body’s production of sex hormones (which in many cases is a permanent short circuit).
A variety of different GnRH over-activators are sold, such as Decapeptyl (Triptorelin), Lupron (Leuprorelin), Suprefact (Buserelin), Synarel (Nafarelin), Zoladex (Goserelin). Since Lupron is the most commonly used one, henceforth, I will only discuss it, but much of what I will say about Lupron also applies to the others as well.
Note: there are also numerous similar drugs which instead temporarily shut down hormone production by directly blocking the GnRH receptor (e.g., Orilissa). Additionally, there are other GnRH over-activators which are only used in animals and have similar side effects to those observed in humans.
Since testosterone fuels the growth of prostate cancer, there was a lot of research on cutting of the body’s testosterone to treat it. Initially the most promising approach was to counteract testosterone with an estrogen analog (DES) which was eventually pulled from the market because it caused a wide variety of issues (e.g., heart attacks, female cancers, and a variety of severe problems in the children of mothers who took DES—which has led many to argue the COVID-19 vaccines may become “the new DES”).
Since Lupron, by burning out GnRH receptors, chemically castrates males (and thereby eliminates their testosterone), a 1984 study was conducted comparing the use of DES to Lupron for patients with prostate cancer which had metastasized to the bones and was hence likely to be fatal. It found Lupron slightly increased their survival rate (although half still were dead within two years of starting the therapy) and it had a slightly different mix of severe symptoms when compared to DES, which in turn was used to argue it was a viable alternative to DES.
When the FDA reviewed this study, the reviewers noted the study had a variety of serious issues so it was difficult to draw any firm conclusions from it. As a result (despite the FDA knowing Lupron had real longterm risks that had not been investigated and other critical aspects of the drug like how the body metabolizes it remaining unknown to this day), Lupron was approved in 1985 as a “palliative treatment of advanced prostate cancer,” a situation which is frequently so debilitating and painful for cancer patients, anything which could potentially somewhat improve it is viewed as justified.
Note: six months ago, Scott Adams, who had advanced prostate cancer, shocked the online community by saying the torture of it had made him decide upon committing suicide in a few months after an important life event had passed—providing a clear example of how dire “advanced prostate cancer” can be.
Since that time, Lupron’s approval was never updated. For those interested, a detailed explanation of why that approval was overtly fraudulent and unwarranted can be found here.
Note: in addition to Lupron offering a very small survival benefit, a strong case can be made that since it is frequently observed to cause a variety of severe complications (e.g., a large increase in fatal heart attacks or diabetes), its reduction in the prostate cancer death rate is actually an artifact of it killing the patients in another manner before a slow growing prostate cancer would. This perspective for example was shared by the Vice President and Chief Scientific Officer of the American Cancer Society.
Once Lupron was approved, its use transitioned from only the most severe prostate cancers to all of them (even though, as shown by a 2009 study of 19,271 men, using Lupron actually increased the death rate). At the same time, a variety of other copycat drugs entered the market. The FDA in turn approved them (or Lupron) for advanced prostate cancer, advanced breast cancer, endometriois (along with its pretreatment prior to surgery), the pretreatment of fibroids before surgery, and preventing precocious (early) puberty.
Note: while I believe the risks of these treatments greatly exceed their benefits, it is also true that a subset of patients exist with those conditions who benefit from Lupron and suffered minimal side effects from the drug.
Additionally, a variety of other off-label uses were concocted, such as:
• “Treating” every imaginable gynecological problem (e.g., large fibroids, difficult menstrual cycles, ovarian cysts).
• In-vitro-fertilization and egg donation protocols.
Note: many young women are paid thousands of dollars to donate their eggs. Unfortunately, a portion of those donors suffer significant complications they are not warned about beforehand and then are left on their own to address. This is likely in part due to the fact Lupron is frequently part of the protocol. Likewise, significant birth defects (which Lupron has been shown to cause in the majority of pregnancies) are frequently reported following IVF—which may explain why despite Lupron being originally patented as a fertility medicine, it could never be formally approved for that use.
• Chemical castration for sex offenders (e.g., pedophiles).
• Helping children become taller (by delaying puberty so their growth plates take longer to close).
• Preventing puberty in a transgendered youth
Note: a more detailed list of the off-label uses can be found here. It is truly remarkable how many different tactics were used to seed these additional uses (e.g., bribing countless doctors and medical charities to promote these drugs) and likewise how many other uses (e.g., for Alzheimer’s disease and Autism) came very close to becoming off-label uses as well.
In turn, there are three important things to take away from all of this.
1. While these drugs were initially developed for men (i.e., prostate cancer), they are frequently given off-label to women. This for example is why Lupron’s FDA insert states its only indication is for the palliative treatment of advanced prostate cancer but it simultaneously warns against pregnant women taking it (even though it’s also used for egg harvesting)
2. Despite having been on the market for decades, there is very little evidence to show these drugs actually benefit those who take them.
3. Given this, along with how incredibly toxic they are (especially to women), it raises a fairly simple question—why on earth are these drugs so popular?
Selling Lupron
Lupron’s manufacturer was stuck with a rather large challenge—how could they got doctors to begin prescribing an incredibly dangerous and ineffective drug? This in turn was accomplished through one of the most overt acts of physician bribery I’ve seen in American medicine.
Since Lupron initially did not sell well, Lupron’s manufacturer took advantage of the existing “standard” which allows chemotherapy drugs to be sold for a very high price and be “forgiven” for their extreme toxicity. This was done by reformulating Lupron into a long acting monthly shot urologists could directly administer to their (prostate cancer) patients and hence directly profit from marking up when they resold it (e.g., Medicare paid 1200 dollars per shot—or roughly 2400 in today’s dollars, and in many cases urologists charged far more, all of which allowed many urologists to make hundreds of thousands of dollars per year administering the shots).
Note: TAP frequently advertised to urologists they could make over $100,000 annually selling Lupron and later cited similar figures to OBGYNs.
To further sweeten the deal, Lupron’s manufacturer frequently bribed urologists and gave them free Lupron samples they “resold.” This was illegal—and eventually resulted in a 875 million dollar fine… but no pharmaceutical executives going to prison.
Because Lupron was immensely profitable, more and more urologists jumped on it, and by the late 1990s Lupron treatments were costing almost a billion dollars per year and accounted for 40 percent of all Medicare payments to many urology practices in the late 1990s. To address this, in 2001, Medicare clamped down on urologists reselling discounted Lupron and in 2003 Medicare lowered the reimbursement for Lupron. In turn from 2003-2005, the rate of inappropriate use of hormonal treatment for prostate cancer dropped from 38.7% to 25.7% and many urologists at the time reported their income had been halved.
Note: one survey found 53% of the urologists who did not believe prescribing Lupron benefitted certain prostate cancer patients still prescribed the drug to them.
This Medicare crackdown on excessive Lupron prescribing for prostate cancer created a major problem for the industry. “Fortunately,” since Lupron was so profitable, many other specialities appeared eager to jump on the Lupron bandwagon, particularly OBGYNs (despite the existing data on using Lupron for gynecological conditions being very poor and in many cases overtly fraudulent). This in turn led to a rapid proliferation of new off-label “uses” for the drug, such as the ones listed above. Remarkably, despite the fact Lupron has been on the market for decades, it is still extremely expensive.
Lupron hence is a very lucrative drug. However it is unclear to me exactly what the current reimbursement is for it (e.g., when I’ve looked online, many patients said they were billed over 10,000 dollars for a single injection).
A recent article exploring the subject found that puberty blockers can cost tens of thousand dollars per year. While insurance typically covers these drugs around 72% of the time, without insurance, according to one source, they cost $4,000–$25,000 per year and according to another source a 3 month Lupron injection is $9500 while a competing 3 month option (histrelin) is $39,000.
Similarly, a 2022 NPR article detailing a man’s prostate cancer experience (where he was given unwarranted Lupron shots) reports he was charged $35,414 for the first shot and $38,398 for the second by a Chicago “non-profit” hospital, and after two years of haggling, was forced to pay the $7,000 not covered by his health insurance.
Let’s compare that to how much Lupron costs (this table designates the average wholesale price pharmacies pay for drugs):
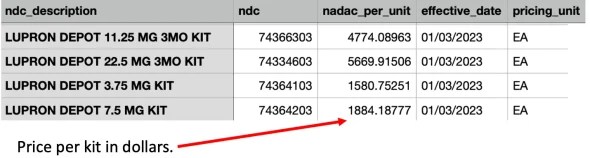
Note: these costs are unusual as they are much higher than what pharmacies typically pay for a drug (especially an older one). The above table is from 2023, and just a year later in 2024, the cost of Lupron went up almost 10%.
Since all of this demonstrates that Lupron is marked up by 5-10 times its original cost when it is resold to patients, I would argue that those who provide these medications may have an ulterior motive in giving them to patients which frequently causes the drugs to be inappropriately prescribed.
Note: one of the most common stories I hear reported from Lupron victims is a tendency for doctors to gaslight them and insist their myriad of health problems could not have come from Lupron, hence making one of their greatest challenges be finding a doctor who can actually help them (or say qualify them for disability since they’ve lost the ability to work). I believe this is partly due to the unusual nature of their injuries and because many doctors have a direct personal investment in believing Lupron is safe and effective (as they aggressively pushed it on their patients—for instance many reported the doctor saying “are you brave enough to try Lupron?”).
Lupron Lawsuits
A curious reality exists with these drugs. To quote Wikipedia:
GnRH analogues [e.g., Lupron] are available as generic medications. Despite this, they continue to be very expensive.
This I attribute both to doctors being heavily incentivized to directly sell these drugs to their patients (rather than cheaper ones made by competitors) and the legal costs associated with producing them.
Since Lupron is so toxic, it had a very high rate of users who were severely and permanently incapacitated by the medication, and hence were willing to go through the arduous process of going to court over it. Since it often took years for the most severe injuries to emerge, this both allowed Lupron’s manufacturer to have the money in place to fight each lawsuit and simultaneously to argue that each injury could not have been related to Lupron. Furthermore, since the legal risk of manufacturing Lupron was so high, I suspect that it scared many competitors away from entering the market as there was a significant barrier towards having enough sales to be able to afford to squash each lawsuit which came along.
In turn, numerous lawsuits have been filed against Lupron’s manufacturer and the doctors who prescribed it, but while some were settled out of court, none to my knowledge were successful, which is extraordinary given that many of the cases revolved about Lupron being used for an experimental (unapproved) use, it causing clear harm to the patient, and it being inappropriately dosed or monitored by the physician (who instead just wanted to give the highly lucrative single injections).
The general sense I have gotten from talking to people injured by Lupron is that they believe Lupron’s manufacturer spent so much on legal defense (e.g., by paying off judges, having the best lawyers or buying gag orders in settlements) that it’s a lost cause to file a Lupron lawsuit regardless of how severe one’s injuries were. In turn, many people have shared that they have been unable to find attorneys who are willing take their case.
Note: one of the things we all found remarkable during COVID-19 was how differently the use of “off-label” prescriptions was treated by our authorities. Despite no injuries occurring, nor any money being made, many of the doctors who saved many lives by prescribing ivermectin or hydroxycholorquine were accused of exploiting their patients and faced harsh penalties for their prescriptions (e.g., Meryl Nass lost her medical license).
Lupron Toxicity
I have had a longtime interest in understanding how pharmaceuticals injure people, so I frequently spend lots of time reading through support groups for people who have been injured by them. From this exploration, I have come to the perspective Lupron is one of the most dangerous drugs on the market due to the sheer volume of injuries patients report, how severe the injuries are and just how much many of them are suffering (e.g., many of these reports are comparable both in their severity and variability to COVID vaccine injuries).
Note: in the late 1990s, a lot of public pressure was building against Lupron, and one group, the National Lupron Victims Network came to prominence as a hub for collecting the evidence of Lupron’s harm and advocating against its continued use. Remarkably, in 2000, shortly before the group was supposed to publish all the data it had collected from surveys on the harms of Lupron, without explanation, it suddenly disappeared. This again illustrates just how far Lupron’s manufacturer went to protect their drug.
Within the Lupron support groups, I find by far the most commonly injured are women. This is followed by individuals who took the drugs to halt a premature puberty, then men, and finally transgendered individuals (as they are a relatively new market).
Note: many of the people who took the drugs during puberty are now having adverse effects decades later (e.g., as discussed in this Kaiser Foundation article). This had led me to suspect the same thing will be “discovered” in the years to come for the transgendered children our society has recently started putting on puberty blockers. Remarkably, a 2009 specialist review of using drugs like Lupron for early puberty or making children taller found “few controlled prospective studies have been performed… and [like now], many conclusions rely in part on collective expert opinion.”
Some of the most commonly reported side effects of Lupron include:
• Numerous studies have found Lupron given at all ages significantly decreases bone density (e.g., many have reported between a 3-10% loss occurring after just 6 months) which often results in fractures (e.g., see this study). Bone loss, in turn, is one of the most commonly reported side effects of Lupron (e.g., many young women report having “bones like an 80 year old,” chronic dental or jaw issues like teeth cracking apart and repeatedly developing unexpected fractures from minor stressors). While this bone loss is often rapid, in many cases, it emerges years after receiving Lupron (e.g., women who went on it during puberty in their 30s learn about it because of how quickly the teeth in their mouth are decaying and being told they are not that far from needing dentures).
• Longterm or permanent damage to female menstrual cycles. For example, Lupron’s clinical trials (revealed through ligation) showed that 62.5% of study subjects had failed to return to baseline ovarian function one year after stopping Lupron (which means, contrary to the manufacturer’s claim, the drugs is not reversible once it is stopped). Many other hormonal issues are also frequently reported (e.g., permanent weight gain, painful and abnormal menses, severe hot flashes and vaginal atrophy)
Note: enlarged ovaries and ovarian pain is a commonly reported symptom of Lupron usage (particularly after egg donation protocols) and there is some data which suggests Lupron causes polycystic ovarian syndrome.
• Sexual dysfunction is commonly reported. For example, one study found 80% of males using these drugs reported being impotent, while another found a 267% increase in impotence was observed after one year of treatment, and another evaluation of a related drug found sexual desire, sexual interest and sexual intercourse were totally annulled. Likewise, chronic pelvic pain (in women), a wide range of chronic bladder issues (e.g., incontinence, bladder spasms, urinary retention, or recurrent UTIs), and testicular pain or atrophy.
Note: these sexual side effect are particularly noteworthy given that in many cases, prostate cancer patients are put onto Lupron for years.
• A variety of psychiatric conditions commonly follow Lupron usage (e.g., a 2002 study of more than 3,000 women on it found 35.5% reported depression). Some of the effects I commonly see reported include anxiety, severe mood fluctuations, major dysphoria, burning rage, suicidality (which sometimes requires being placed on a suicide watch), and losing the ability to function in social situations.
Note: particularly in transgender or precocious puberty groups, users report a profound loss of identity or personality changes, feeling “like a stranger in my own body” due to hormonal suppression. Likewise, many users report frequently describe social withdrawal, inability to maintain relationships, or marital strain due to Lupron’s emotional and physical toll (e.g., “Lupron killed my marriage; I wasn’t myself). Finally, many users report losing their jobs and financial stability due to being disabled by Lupron.
• Cognitive dysfunction (e.g., brain fog or memory loss) is also frequently reported. One study found evaluating women receiving IVF found 72% showed difficulty with memory while on Lupron, some subjects had significant cognitive deficits, and 11% showed very substantial neurocognitive issues.
• Online, children and young women treated with Lupron often report seizures or convulsions, tinnitus (and other hearing issues), visual disturbances (e.g., floaters, blurred vision, or photophobia). Hormonal issues besides those with the sex hormones are also frequently reported such as thyroid issues (e.g., hypothyroidism and goiter), adrenal issues (e.g., extreme fatigue, salt cravings, or low cortisol) and diabetes or glucose dysregulation. In many cases, these onset immediately after starting Lupron (with no prior signs of them) and are then permanent.
• IQ loss in children (e.g., one study found a 7 point drop, while another found an 8 point drop).
• Lupron (and related therapies) are associated with a variety of different heart conditions, as Lupron (when used for prostate cancer), according to one paper, appears caused a 10–50% increase in the risks of coronary heart disease, myocardial infarction, strokes and sudden cardiac death (e.g., this study the paper referenced shows a massive increase in heart attacks). Many other concerning heart conditions have also been linked to Lupron and numerous medical textbooks explicitly warn about them. The FDA in turn eventually issued a warning in 2010 about this increase risk of heart problems (and diabetes) in men and acknowledged that no research existed to assess those risks in women or children.
• A wide range of gastrointestinal disorders (e.g. severe abdominal pain, irritable bowel syndrome, or growths that require excisions) and genitourinary (e.g., frequent urination, incontinence and interstitial cystitis) are frequently reported. Many of these likely result from Lupron disrupting the autonomic nervous system and it cutting off blood flow to tissues of the body, which for example is why it shrinks fibroids.
Note: originally, the FDA was extremely concerned about the potential harm which could result from Lupron cutting off the blood flow to critical organs.
• Many Lupron patients report crippling joint pain and severe (early) arthritis. For example, a study of more than 3000 women found that 76.7% reported joint pain. Likewise, severe pain throughout the body, tendon issues (e.g., tendinitis, severe pain in the tendon, or tendon ruptures), muscle wasting or pain and degenerative disc disease is often reported in support forums.
Note: many of these symptoms overlap with what’s commonly reported by patients with ligamentous laxity (e.g., they are hypermobile and have frequently cracking joints). I recently wrote an article detailing how hypermobility is a common characteristic of sensitive patients and its common association with manganese deficiency.
• Immune suppression (e.g., within the bone marrow) and a wide range of severe autoimmune conditions (e.g., Sjogren’s, lupus and various thyroid conditions) are frequently reported by Lupron patients. Chronic skin conditions (eczema, psoriasis, or chronic rashes) not responsive to treatment and significant hair loss are also reported. Finally, other more severe immune-related side effects such as unusual tumors developing (or rapid growth of an existing one), anaphylaxis are also reported (along with other organ dysfunctions like elevated liver numbers).
What should jump out from this list is how frequent, severe, and wide-reaching these injuries are. This in turn helps to explain why the FDA’s system for reporting drug injuries (which catches 1-10% of those which occur) has received 76,221 Lupron injury reports, of which 41,895 were severe and 11,917 were fatal. Likewise, consider how frequently a myriad of conditions occurred when Lupron was tested in men who had prostate cancer (per Lupron’s FDA package insert):
Anemia (6.6%), Asthenia (7.4-12.2%), Back Pain (5.3%), Blood in Urine (6.6%), Constipation (9.9%), COPD (5.3%), Coronary Heart Disease/Angina (5.3%) Cough (6.6%), Dehydration (8.2%), Dizziness/Vertigo (5.3-6.4%) Edema (5.3-8.2%) Elevated Blood Pressure (6.6%) Fatigue (13.2%) Flu Syndrome (12.2%) General Pain (23.2-32.7%) GI disorders (10.2-16%), Headache (6.4-10.2%), Hot flashes/sweats (46.9-58.9%) Impotence (5.4%), Infection (5.4%), Injection Site Reaction (8.2-19.2%) Insomnia/Sleep Disorder (8.6%), Insomnia/Sleep Disorders (8.5%) Joint Disorders (11.7-16.3%) Joint Pain (9.3%) Libido decreased (5.4%) Muscle Pain (7.9-8.2%) Neuromuscular Disorders (6.1-9.6%) New Cancer (7.3%) Pain While Urinating (6%) Paresthesia (8.2%) Rash (6.6%), Respiratory disorder (6.4-10.7%) Shortness of Breath (5.3%) Skin Reactions (8.5-12.2%) Testicular atrophy (5.4-20.2%) Urinary disorder (12.2-14.9%) Urinary Tract Infection (6%).
Likewise, this is what the FDA reports occurred when Lupron was tested on women for endometriosis:
Acne 10%, Altered Bowel Function (constipation, diarrhea) 14%, Asthenia 8-18%, Breast Changes/Pain/Tenderness 6%, Breast changes/tenderness/pain 6%, Decreased libido 10-11%, Depression/emotional lability 11-31%, Dizziness/Vertigo 11-16%, Edema 5-7%, General pain 8-24%, GI disturbances 7%, Headache 26-65%, Hot flashes/sweats 73-98%, Insomnia/Sleep Disorder 31%, Joint disorder 8%, Memory Disorder 6%, Nausea/vomiting 5-25%, Nervousness/Anxiety 5-8%, Neuromuscular disorders 7%, Paresthesias 7%, Skin reactions 10%, Vaginitis 11-28%, Weight gain/loss 12-13%
Unfortunately, while the above list is terrible (particularly given that the “benefit” of the Lupron in both cases was minimal at best), it should be noted that:
• Pharmaceutical companies always conceal adverse events which occur in their trials.
• This list only includes conditions more than 5% of trial recipients developed while on the drug. In turn, a variety of rarer but much more severe conditions did not make this list.
• This list was not evaluating the long-term effects of Lupron (which are typically the most severe).
Because of how toxic Lupron is, by far the most challenging part of this article was accurately synopsizing the thousands of injury reports I’ve read over the years (as I felt their heart wrenching stories deserved to be heard but simultaneously, there are just far too many for me to fit into any number of articles here).
Generally speaking, Lupron (like the COVID vaccines) causes the body to age prematurely—which in the case of Lupron provides an important insight on the importance of hormones as these victims provide a unique insight into what happens as the body loses those essential messengers (something which also occurs with age). This why in addition to profound bone loss, Lupron also frequently causes other degenerative processes like hairloss, vaginal atrophy, receding gums, and declining vision.
For each of those symptoms (and many others), I’ve read countless testimonials describing the anguish of having their body rapidly age in front of their eyes and the general despair that accompanies decades of suffering with these ailments and the fact there is no one who will help them.
Additinally, one of the most common stories I hear in the support groups are women who profoundly regret taking it for endometriosis as beyond it permanently debilitating them, it frequently did not help (or worsened) their endometriosis.
Note: endometriosis is another condition which is poorly treated by the medical system. Typically the best option within the conventional paradigm is to have it be surgically removed, but unfortunately, there a very few surgeons competent surgeons who do this (e.g., the person we use is an 8 hour drive away from us) and there is also a surprising lack of knowledge within the OBGYN field of how to appropriately manage endometriosis.
Like the COVID vaccine injured, many of those injured by Lupron report not a few, but rather dozens of debilitating symptom. Furthermore, there is often a significant overlap in these symptoms (e.g., both frequently experience fibromyalgia, severe neuropathies, chronic fatigue, headaches, insomnia migraines, hypersensitivities to everything, seizures, and lightheadedness or fainting).
Lupron Stories
Since there are so many reports of people being harmed by Lupron, it’s impossible for me to ever do justice to their experiences in a brief article. As such, I will simply quote ten of them with the caveat they are only the tip of the iceberg.
Within 2 weeks of starting Lupron therapy [for endometriosis], I was walking with sticks due to the pain in my hips and ankles. I stopped eating. My skin was dry, flaking and itchy. I had no short term memory & my concentration got so bad I couldn’t safely drive. I didn’t sleep a wink for months. I was so depressed that I stayed in bed for days at a time.
I’m a calm, sane person. I’m not kidding, that sh*t made me feel insane [taken following endometriosis surgery]. A horrible emotional roller coaster. If I had to do it over again, I’d have just had the hysterectomy.
Anyone else have terrible mood changes with Lupron [for IVF]… My mood is all over the place… I hated lupron. I had terrible headaches and severe joint pain. Just felt crummy all around.
The side effects from my lupron injection [for prostate cancer] is awful compared to my radiation treatment… frequent urination, bowel discomfort, and a rash… but the Lupron is making me feel like I’m hit by a bus!! Chills, fever, runs, aching body, emotional.
I had a terrible time with Lupron when I used it as endo treatment ten years ago: hot flashes, night sweats, weird hairs growing everywhere, headaches, mood issues… I ended up only doing five of the six months because I felt so badly.
Lupron for me stopped the endo pain… But yeah, it replaced it with severe joint pain, hot flashes, weight gain and mood swings… I’ve since had a hysterectomy and am going through all of that again from real menopause.
I had lupron shots as part of my IVF protocol. I don’t exactly regret it… but I wish there had been another way… it caused the worst fibromyalgia flare of my life! I ended up in the ER and couldn’t walk for a few weeks. I’ve never experienced so much pain in my life.
The side effects were PROFOUND and BRUTAL [was taken for breast cancer]… Giving Lupron to a healthy minor? It should be criminal.
Lupron depot horror... I’m official a year out from my 3 month use… it’s given horrible life long side effects and other health conditions… It’s horrible!! It does not help shrink endo growths… wrecked havoc on my health.
I took Lupron, the original puberty blocker, for endometriosis. Before Lupron I only had endometriosis. After Lupron I have bone death in both hips, brittle bones that break easy, multiple fractures in feet, hypothyroidism, and a non cancerous pituitary tumor.
So, as you might expect, individuals who took them as children did not have the best experiences either:
As a parent of a child who went through precocious puberty and was given puberty blockers (Lupron), I watched my healthy mentally stable son fall into severe depression, multiple suicide hotline contacts including a visit from emergency services. Self harm scars and self isolation. He began to question his sexuality and gained excessive weight. All before 12
My son had one Lupron shot for precocious puberty at age 4—the side effects were horrible: aggression, pain, and now years later, we’re seeing bone density issues and growth problems. It’s a nightmare we regret starting.
My daughter took Lupron for precocious puberty starting at age 7. Now at 25, she has degenerative discs in her spine, chronic joint pain, and hypothyroidism. We thought it would help her grow taller, but it’s caused lifelong hell—no one warned us about the bone death and fractures.
Valerie Ward, 25, who lives outside of Pittsburgh, said she took Lupron for precocious puberty, from age 9 to 12. Like Derricott, Ward said she sees a carousel of medical specialists for excruciating muscle and bone pain, depression, weakness and fatigue.
Put on Lupron at 10 for precocious puberty to ‘buy time for height,’ but it worked too well—stopped puberty entirely, then I needed growth hormone shots for years. Now 30, flat-chested with endo and weak bones; wish we’d never done it.
I was given Lupron as a child for precocious puberty. Now in my 30s, my bones are like an 80-year-old’s—brittle, fracturing from nothing, teeth crumbling. Doctors said it was safe, but it’s ruined my life [this is a paraphrase summary of many posts by this user].
My son was put on Lupron at 9 and we were NOT told bone damage was a potential side effect. Today, at 24 he has severe osteoporosis and the bones of a 75 year old!! Even this was discovered by happenstance. Trying to get help with this condition has been nearly impossible.
I was on Lupron for a 9 months in 1995, fast-forward 30 years I now have full blown osteoporosis from the lupron! I break bones every other week! No child should be taking any of it, I don’t have a problem when you’re an adult and you know the consequences but children no!”
WPATH’s Transgender “Guidelines”
Evidence based medicine was created so that harmful and irrational dogmas within the medical field could be overturned by scientific evidence proving there was no justification for doing them. While this was initially helpful, the process gradually became corrupted as the pharmaceutical industry realized doctors could be made to believe only the “best” evidence should be trusted, and the groups purveying the “best” available evidence (e.g., the premier medical journals) could be easily bought out.
A key part of the push to buy out the “best” evidence has been to create authoritative guideline committees who are tasked with evaluating the existing scientific evidence and coming to a consensus over what constitutes the best practice of medicine—a process which is fairly easy to corrupt since the industry can simply pay off each member of the “expert” committee.
This for example is why Anthony Fauci was allowed to appoint the members of the government committee which decided the standard of care for COVID-19 and Fauci chose individuals who were both his friends and had significant financial ties to his pet drug Remdesivir. In turn, that committee concluded only the extremely expensive COVID-19 treatments (e.g., remdesivir—which was repeatedly shown to worsen rather than improve COVID-19) should be used to treat COVID-19, whereas the safe and effective (but non-commercializable) therapies (e.g., ivermectin) were never allowed into the treatment guidelines despite dozens of trials from around the world proving they worked.
Note: corrupt committees are a recurring problem. For example, the government committee which created the statin usage guidelines we all follow that erroneously concluded everyone needed to be on the statins was filled with people taking money from the statin industry.
In the field of transgendered medicine, much of what is being done is a result of physicians following the existing guidelines that have been created by the World Professional Association for Transgender Health (WPATH). For this article, I reviewed exactly what their guidelines had to say about giving puberty blockers (GnRH analogs) like Lupron to children.
First, they strongly endorsed administering these drugs:
• The moment transgender children begin the earliest signs of puberty as this provides a greater benefit that administering them later on.
• As a stopgap measure for children who have mostly gone through puberty and are considering starting opposite sex hormones but are not yet sure they wish to begin hormone therapy (e.g., due to a disagreement with their parents over doing it).
• For adolescents who are distressed by their body’s menstrual cycles (since the blockers stop menstruation) or penile erections since Lupron suppresses both of them. This is similar to how the guidelines emphatically cite the benefit of these drugs creating “a vast reduction in the level of distress stemming from physical changes that occur when endogenous puberty begins.”
• To help males hoping to achieve a female’s hormone levels do so (as Lupron and its ilk suppress testosterone).
Note: they also acknowledge there are other “individualized” circumstances where someone who has completed puberty may benefit from these drugs.
Second, they advise against using them when:
• The child and their family cannot attain or afford them (in which case specific hormones like progestins are instead used).
• Prior to the earliest signs of puberty. This is because it can potentially interrupt a critical part of their psychological sexual development (however, this logic only applies to very start of puberty and not the rest of it). They do however advise regularly monitoring these children to detect when they start puberty so the blockers can be immediately initiated and provide for a few exceptions where the drugs can be administered prior to the start of puberty.
Third, while repeatedly claiming these drugs are safe and their effects are rapidly reversible, they do lightly acknowledge a few issues might exist.
Note: feel free to skim this section—I wrote it because I felt it was important to accurately depict every single “warning” WPATH provided against these drugs.
General:
- “[The use of puberty blockers] is generally safe with the development of hypertension being the only short-term adverse event reported in the literature.”
Bones:
- “While GnRH analogs have been shown to be safe when used for the treatment of precocious puberty, there are concerns delaying exposure to sex hormones (endogenous or exogenous) at a time of peak bone mineralization may lead to decreased bone mineral density. The potential decrease in bone mineral density as well as the clinical significance of any decrease requires continued study.”
- “For adolescents older than 14 years, there are currently no data to inform HCPs whether GnRHas can be administered as monotherapy (and for what duration) without posing a significant risk to skeletal health.
- “The rate of bone mineralization, which decreases during treatment with GnRHa’s, rapidly recovers.”
- “Based on scientific evidence currently available examining the use of GnRH agonists in transgender adolescents, it is unclear whether or not using puberty blockers in adolescence will increase the risk for future fractures in transgender adults.”
- “[They] can result in osteoporosis if doses of estrogen given concurrently are insufficient.”
- “A prolonged hypogonadal state in adolescence…due to..iatrogenic causes such as GnRHa monotherapy..is often associated with an increased risk of poor bone health later in life. However, bone mass accrual is a multifactorial process that involves a complex interplay between endocrine, genetic, and lifestyle factors [so] all contributing factors should be considered [and] a multidisciplinary team and an ongoing clinical relationship with the adolescent and the family should be maintained when initiating GnRHa treatment.”
Fertility:
- They “may also result in menstrual suppression.”
- “GnRHas may also be used for menstrual suppression. GnRHas impact the maturation of gametes but do not cause permanent damage to gonadal function. Thus, if GnRHas are discontinued, oocyte maturation would be expected to resume.”
- “GnRHas inhibit spermatogenesis. Data suggest discontinuation of treatment results in a re-initiation of spermatogenesis, although this may take at least 3 months and most likely longer.”
- “Pubertal suppression and hormone treatment with sex steroid hormones may have potential adverse effects on a person’s future fertility [thus] the potential implications of the treatment and fertility preservation options should be reviewed by the hormone prescriber and discussed with the person seeking these therapies.”
Adversely impacting a gender transition:
- The potential negative psychosocial implications of not initiating puberty with peers may place additional stress on gender diverse youth, although this has not been explicitly studied.”
- “Treating an TGD adolescent with functioning testes in the early stages of puberty with a GnRHa not only pauses maturation of germ cells but will also maintains the penis in a prepubertal size. This will likely impact surgical considerations if that person eventually undergoes a penile-inversion vaginoplasty as there will be less penile tissue to work with. In these cases, there is an increased likelihood a vaginoplasty will require a more complex surgical procedure, e.g., intestinal vaginoplasty.”
Hopefully, as the previous section showed, WPATH’s depictions of the dangers of these drugs (Lupron etc.) is highly misleading as a large body of evidence exists which overtly contradicts what WPATH put forward. Given that I was able to compile that evidence in under a week, it is surprising a team of “experts” who have spent years working to produce these guidelines were unaware that literature (and likely much more) existed. In turn, because doctors are trained to trust guidelines, they assume that since WPATH said puberty blockers are “safe and effective” they indeed are, hence leading to them aggressively pushing them on patients and gaslighting anyone who reports side effects from them.
Furthermore, the thing I found the most remarkable about WPATH’s guidelines was that while they were unaware of the dangers of Lupron (and its related drugs), they repeatedly referenced certain dangers of giving specific hormones, and in numerous cases characterized the Lupron as safe and effective alternative to the more dangerous hormone therapy. I in turn suspected this is because the blockers cost far more than artificial hormones, and once administered, often require the lifelong purchase of artificial hormones (e.g., to prevent some of bone loss and to make up for the body no longer producing its natural hormones).
All of this led me to believe that like many before them, those involved in writing these guidelines (and some of the authors they referenced) were paid off to promote Lupron and its ilk, but as I have not had the time to do the investigation to confirm this, I can’t state it with certainty.
The WPATH Leaks
Since WPATH has continually publicly advocated for transgender care to be made available to everyone that organization has received increasing scrutiny from the public.
Recently, this resulted in internal documents and correspondences from WPATH being leaked. I reviewed those leaks to see exactly what WPATH’s members were saying in private about puberty blockers. From reviewing all of it, I learned that much like each other group which has promoted the off-label usage of Lupron, WPATH was:
• Not entirely sure what the long-term consequences of this push for those drugs was and in essence, much of what they were doing was a large experiment.
• Recognized that a variety of significant side effects would occur in children who took the blockers (e.g., some would permanently lose their libido or the ability to have an orgasm and many children would lose the necessary emotional developmental process that occurs during puberty).
• Despite continually claiming otherwise publicly, they knew the effects of Lupron were often not reversible.
• Recognized that the children they were giving the blockers to were too young to fully comprehend the dangers of these drugs but nonetheless were seeking to initiate their use as early as possible.
It’s relevant at this point to note that the puberty suppression experiment began because transgender adult males were dissatisfied with the results of their medical transition because they did not “pass” well as women due to a “never disappearing masculine appearance.” Therefore, the Dutch researchers came up with the idea to use gonadotropin-releasing hormone agonists (GnRHa) to block the testosterone surge of male puberty in the hopes of achieving more feminine appearances in adulthood. The increased risk of false positives due to early intervention was noted, but the cosmetic advantages to adult natal males who identify as women were deemed more important.
Note: WPATH members also routinely discussed puberty blockers being administered to developmentally delayed children (e.g., those with autism), who due to their conditions had an even greater inability to consent to these drugs. This dovetails into another uncomfortable fact rarely discussed in the transgender debate—vaccination significantly increases the likelihood of transgenderism, most likely because autistic individuals are receptive to messaging which tells them the disconnect they feel with their body and society is not due to vaccine brain damage but rather tothem being the wrong gender—all of which I detailed extensively here.
Given all the things I’ve seen the pharmaceutical industry repeatedly do to make money during my lifetime, very few things surprise me these days. Nonetheless, even I was a bit taken aback when I discovered through these documents that there has been a push to affirm “plural identities” (multiple personalities) within WPATH. In turn, there are numerous cases which have been presented at WPATH conferences (e.g., under the umbrella of UCSF—one of America’s premier medical institutions) where each personality of an individual with split personalities was assessed for its sentiments on beginning a gender transition and at least one instance where some of the personalities did not consent but the transition was nonetheless deemed “ethical” and proceeded.
Note: I have compiled numerous cases which demonstrate that the one consistent principle in medical ethics is that whatever makes money will inevitably been seen as the “ethical” choice.
In short, given all of this, I strongly suspect WPATH (and possibly other members of the industry) were paid off to ensure puberty blockers would be a mainstay transgender treatment—particularly since you can often map out multi-year if not multi-decade campaigns done by pharmaceutical companies to ensure vibrant and continually growing markets for their products (e.g., the completely unjustifiable COVID lockdowns made many so desperate for a solution they eagerly embraced the vaccine without critically thinking about its multitude of red flags).
Note: much of the modern transgender push in medicine resulted from a provision in Obamacare (along with regulatory decisions from the Obama and Biden administrations) which mandated insurance companies provide coverage for gender transitions (which included paying for the costly GnRH agonists).
Conclusion
When you consider the entire Lupron saga, it is truly remarkable that a drug this dangerous has managed to stay on the market for decades, particularly given that it still demands an exorbitant price despite there being numerous significantly cheaper generic formulations which could be used instead. Even more remarkable is the fact that there is no evidence to support most of the things its used for, and now almost 40 years later, that the FDA has still not updated its 1985 approval.
Consider for a moment the contrast with what we saw during COVID-19, where numerous widely used (and widely recognized to be safe) drugs were effectively banned in the treatment of COVID-19, despite no viable therapy existing for the illness, many reporting dramatic improvement from those protocols and widespread public pressure for these off-patent drugs to be used to treat COVID. In contrast, the FDA has ignored decades of complaints and evidence hormone blockers severely injure patients, and despite widespread public outcry against their use, has used countless clinics to routinely prescribe them in an experimental manner they were never approved for. In short, Lupron represents a classic case of where the FDA has a statutory obligation to prohibit the reckless off-label use of this drug, yet has never done so due to the immense money being made from it and the multi-decade campaign to ensure it has a vast sales market.
Likewise, the Lupron situation is analogous to what we are seeing with the COVID-19 vaccines. Like Lupron, they are extraordinarily toxic and in 1-2 injections, often permanently destroy someone’s health in a myriad of ways—but nonetheless are relentlessly defended by the FDA.
In my eyes, the one bright side to the COVID debacle was that the sheer egregiousness of it (mandating an experimental, dangerous and ineffective vaccine while simultaneously suppressing numerous safe and effective treatments for the disease) opened many people’s eyes to the rot within our healthcare system. In turn, people are now seriously open to ideas like how many young women were severely injured by the Gardasil (HPV) vaccine, the century of evidence that childhood vaccines cause sudden infant deaths, or the notion the vaccines cause autism.
Like Lupron, the people who have suffered from those previous vaccines had done everything they could for decades to alert the public to how dangerous they were, but by and large, their pleas had fallen on deaf ears. However, in the same way the COVID-19 vaccines became heavily politicized (which in turn caused half of America to begin seriously scrutinizing all vaccines), the use of Lupron has also become heavily politicized due to the medical industry’s greedy decision to push the drug on our children.
Because of this, we are now seeing leaks (e.g., the recent WPATH one) emerge which are exposing how reckless and unwarranted certain uses of Lupron are. More importantly, since the issue has been politicized, a lot of people are willing to listen and major groups (e.g., numerous Republican states and England’s National Health Service) are now responding to the public pressure and prohibiting this use of these drugs. Similarly, certain states are making it easier to sue doctors who give puberty blockers to children and many lawsuits are now being filed. This in turn is causing the cost of their medical malpractice insurance to skyrocket and in many cases be more than what the doctors can afford, hence is making them be unable to continue giving these drugs to children.
This in turn is what those injured by Lupron had fought for decades to make happen and it is my sincere hope that our newfound public scrutiny on these drugs will make it possible to at last bring awareness to how incredibly harmful its other uses are too. I thank each of you for reading this and your help in bringing awareness to this medical atrocity and everyone (e.g., the forgotten women) who has suffered from those drugs.
‘A Form of Bribery’: FDA, HHS Crack Down on Misleading Drug Ads
By Michael Nevradakis, Ph.D. | The Defender | September 10, 2025
Pharmaceutical companies will be required to provide full safety disclosures in direct-to-consumer (DTC) advertisements of their products, according to a new policy HHS and the FDA announced Tuesday.
DTC advertisements “can mislead the public about the risks and benefits” and “encourage medications over lifestyle changes,” according to a memorandum by President Donald Trump outlining the policy.
The U.S. Food and Drug Administration (FDA) will send nearly 100 “enforcement action letters” and thousands of warning letters to pharmaceutical companies and drug retailers who have “increasingly been promoting drugs with no mention of side effects at all,” FDA Commissioner Marty Makary said in a post on X.
The policy also addresses online pharmacies that promote drugs with “no mention of side effects, and paid social media influencers advertising drugs,” Makary wrote.
Administration officials told ABC News that drugmakers often market their products on social media using influencers who are not clearly identified as paid spokespeople.
Mary Holland, CEO of Children’s Health Defense, called the new policy “a major victory” that will “dramatically increase the price of pharma advertising, discourage uptake because of side effects and make Big Pharma‘s lawyers stay up at night worrying that they may not have adequately disclosed risks.”
“This will greatly contribute to making America healthy again because it will start to dismantle Pharma’s grip on Big Media,” Holland said.
‘Pharmaceutical ads hooked this country on prescription drugs’
In announcing the new policy, the U.S. Department of Health and Human Services (HHS) said the ads have “distorted physician prescribing habits and patient decisions.”
The advertisements use positive emotional appeals to encourage people to get those medications, HHS said.
The new policy stops short of an outright ban on the advertising. Instead, the policy will require DTC advertisements to “report full contraindications, boxed warnings, and common precautions” — a return to regulations in effect until 1997.
HHS said the loosened regulations in place since that year created an “explosion of DTC pharmaceutical advertising,” which led to “public deception from patient confusion” and “patient harm via inappropriate demand for medications and misalignment of therapeutic choices with actual patient needs.”
Administration officials told ABC News the new policy “is the strongest, boldest action we can take to make sure that patients have adequate safety information on pharmaceutical ads.”
They said no additional steps are planned to regulate such ads.
“Pharmaceutical ads hooked this country on prescription drugs,” U.S. Health Secretary Robert F. Kennedy Jr. said in a statement. He added:
“We will shut down that pipeline of deception and require drug companies to disclose all critical safety facts in their advertising.
“Only radical transparency will break the cycle of overmedicalization that drives America’s chronic disease epidemic.”
The new policy was announced on the same day the White House released its Make Our Children Healthy Again strategy report, which states that the federal government “will increase oversight and enforcement under current authorities for violations” of DTC drug advertising laws.
Time reported that the U.S. and New Zealand are the only countries that permit DTC drug ads. According to Digiday, Big Pharma spent $30 billion on advertising in 2024. According to HHS, drugmakers spent $369.8 million in social media advertising in 2020.
Relaxed advertising rules had ‘clear negative impact on public health’
According to the White House memo, the U.S. Congress granted the FDA authority to regulate prescription drug advertising in 1962. DTC drug advertising in the U.S. began in 1981, but regulations were loosened in 1997, resulting in a 330% increase in drug advertising by 2005.
According to HHS, the relaxed regulations permitted drugmakers to direct the public to websites, toll-free phone numbers and package inserts for details on contraindications and common precautions.
An HHS fact sheet states that this “loophole … had a clear negative impact on public health,” contributing to about 31% of the rise in U.S. drug spending since 1997.
According to HHS:
- Patients who consulted with their physician about a DTC-advertised drug were about 17 times more likely to receive a prescription than those who didn’t — the result of persuasive marketing techniques.
- 91% of direct-to-consumer drug ad claims featured social approval as a result of product use and 94% employed positive emotional appeals.
- Prescription drug use among Americans increased from 39% (1988-1994) to 49.9% (2017-2020) in the last 30 years.
Following the FDA’s loosening of its regulations in 1997, the agency’s enforcement actions also decreased. “Enforcement letters plummeted from over 130 annually in the late 1990s to just three in 2023,” according to the fact sheet.
HHS said enforcement actions will intensify, with the issuing of “dozens of enforcement letters related to false and misleading advertising, which makes the drug at issue misbranded.”
The FDA will also “send a letter to every single sponsor of an approved drug or biologic … warning them that the Agency is no longer asleep at the wheel, putting them on notice that FDA will be actively enforcing violations of the law, and directing them to remove all non-compliant promotional materials from the market.”
Drug advertising ‘a form of bribery’
Attempts by the federal government to enact a full ban on DTC drug advertisements are likely to face legal challenges, some legal experts say.
A report by The Lever in January states that it is “relatively unlikely” the federal government will be able to ban DTC pharmaceutical ads, partly because courts have previously rejected such attempts on First Amendment grounds.
Attorney Rick Jaffe wrote last year that while legal precedent exists through the 1970 ban on cigarette advertising in broadcast media in the U.S., “An advertising ban on the entire Pharma industry would be a much heavier lift.”
Despite such obstacles, the End Prescription Drugs Now Act, introduced in June and pending before Congress, would ban DTC prescription drug advertising entirely if passed.
Jeffrey Tucker, president and founder of the Brownstone Institute, said the Trump administration’s new policy is “entirely consistent with the First Amendment but will very likely make vast amounts of existing DTC advertising too arduous for it to continue as is.” He said:
“An outright ban would be easily overturned by the courts on First Amendment grounds. On the other hand, in a free society, every seller of products and services has an obligation to warn of risks. This normal practice has been neglected for a long time. This is what has allowed Pharma to spread its wings without accountability and without ensuring informed consent.
“This is an excellent step, not only to protect the public but to curb Pharma capture of the major media.”
According to CNN, the healthcare and drug industry is fourth among all industries in television advertising expenditure, accounting for 11.1% of the market. Prescription drugs accounted for 30.7% of ad minutes across evening news programs on ABC, CNN, Fox News, MSNBC and NBC last year, according to The Wall Street Journal.
According to a 2019 Forbes report, Pfizer spent twice as much on marketing its products as it did on research.
Last year, the Congressional Budget Office estimated that a 10% increase in DTC advertising results in a 1% to 2.3% increase in consumer drug spending.
Mark Crispin Miller, Ph.D., a professor of media studies at New York University whose research and teaching focus on propaganda, said such expenditures have enabled Big Pharma to exercise significant editorial control over the legacy news media.
Miller said:
“Drug advertising, like all commercial advertising, is a form of bribery that corrupts all media that carry it. This development has been the most destructive of them all. Nothing on TV, radio and/or the Internet should be ‘brought to you by Pfizer’ or any other corporate poisoner.”
This article was originally published by The Defender — Children’s Health Defense’s News & Views Website under Creative Commons license CC BY-NC-ND 4.0. Please consider subscribing to The Defender or donating to Children’s Health Defense.
‘Tough Thing to Defend’: FDA Holds Heated Debate on ‘Untested, Unapproved’ Fluoride Supplements for Kids
By Brenda Baletti, Ph.D. | The Defender | July 24, 2025
The decision whether or not to prescribe unapproved fluoride supplements to children needs to be based on data — “It can’t be done with opinion,” Dr. George Tidmarsh said Wednesday during a public meeting held by the U.S. Food and Drug Administration (FDA) to solicit public input on safety concerns associated with the supplements.
Tidmarsh was tapped this week to lead the FDA Center for Drug Evaluation and Research, which regulates over-the-counter and prescription drugs.
In May, the FDA moved to ban fluoride supplements after a systematic review of recent science by top government scientists, published in January in JAMA Pediatrics, reported that early fluoride exposure was linked to a decrease in children’s IQ scores.
During Wednesday’s meeting, Tidmarsh said:
“That’s a huge issue. Everybody should be concerned about that. What this is saying is that the fluoride in water was causing a cognitive decrease in the younger children. And the randomized studies say that there is no benefit. So that’s a tough thing to defend.”
“Our job here is to use evidence,” he added, citing the recently updated Cochrane Review on fluoride, which found that water fluoridation offered little to no protection against cavities in children.
Although the FDA never approved fluoride supplements, which come in tablets and lozenges, doctors have for decades routinely prescribed them to children — including babies as young as 6 months old — to prevent cavities.
Research has shown for more than two decades that any benefit to teeth from fluoride comes from topical applications — like toothpaste — not from ingesting the drug.
The supplements are known to cause dental fluorosis, a tooth discoloration that is a marker of fluoride overexposure. Overwhelming evidence now shows that ingesting fluoride is linked to lower IQ in children, neurobehavioral issues and thyroid problems.
Fluoride advocates say safety studies exist, but don’t cite them
Wednesday’s day-long meeting, facilitated by the Reagan-Udall Foundation for the FDA, turned contentious at times, as researchers presented evidence of fluoride’s risks to children’s health and debated whether to pull the supplements from the market.
Eighteen speakers argued for and against the supplements, including top researchers on fluoride’s toxic effects. Speakers included Kyla Taylor, Ph.D., an epidemiologist at the National Institutes of Health and an author on the National Toxicology Program’s recent report and JAMA study showing that fluoride exposure for pregnant mothers and infants is linked to lowered IQ among children.
Christine Till, Ph.D., a professor from York University in Toronto and author of a highly cited 2019 study that reported similar findings, shared research on fluoride’s damaging effects on the thyroid.
Commenters also included pro-fluoride lobbyists and advocates, such as Dr. David Krol, with the American Academy of Pediatrics, and Dr. Scott Tomar, with the American Dental Association (ADA).
In his opening remarks, Tidmarsh warned about the need for data in response to presentations by supplement advocates, including Dr. James Bekker, a pediatric dentist and a member of the Utah Dental Association.
Bekker said, “We’ve got to have a balance. We’ve got to have a situation where the right amount of fluoride is present during development. As we look at ways in today’s world of achieving that balance, supplements play a very important role.”
“When we don’t have fluoride, there are certain things that happen that are very disturbing,” Bekker added. “We have an increase in tooth decay, and we have an increase in the use of emergency services to receive care for dental emergencies.”
However, Tidmarsh called out Bekker, comparing the lack of evidence in his presentation with the presentation by Dr. Bill Osmunson, a dentist associated with the Fluoride Action Network (FAN), who pointed out that no randomized controlled studies had ever been conducted on the supplements.
Bekker countered that he didn’t want to overwhelm people with information. “That data is very available. It’s important to understand that,” he said, citing no specific studies.
‘Just take them off the market’
The move to ban the supplements comes on the heels of a federal court decision last September that water fluoridation at current U.S. levels poses an “unreasonable risk” of reduced IQ in children and that the U.S. Environmental Protection Agency (EPA) must take regulatory action to address that risk.
Since then, more than 60 communities and two states — Utah and Florida — have voted to stop adding fluoride to their water. The EPA is appealing the decision.
Water fluoridation has been practiced in the U.S. for decades, long advocated by lobbying organizations like the ADA. The Centers for Disease Control and Prevention (CDC) continues to celebrate it as one of the 10 greatest public health achievements of the 20th century.
As water fluoridation comes under threat, fluoridation advocates have taken up the cause of defending the untested, unapproved supplements.
“Honestly, it’s ridiculous that we are even having this discussion — the supplements have never been tested, they have never been approved, and we know that early fluoride exposure can harm children,” Dr. Griffin Cole, conference chairman of the International Academy of Oral Medicine and Toxicology, told The Defender after the meeting. “Just take them off the market.”
Cole, who presented data on fluoride’s neurotoxicity, said these debates had become tiresome because fluoride advocates repeat the mantra that it is “safe and effective,” and are unresponsive to evidence of fluoride’s harm.
“I can understand why most people who aren’t informed in fluoride science would simply succumb to business as usual,” he said.
Advocates call supplements ‘safe and effective’ despite no safety testing
Dr. Charlotte Lewis, a pediatrician and one of the panelists who defended supplements, was slated to comment on the content of the presentations about fluoride’s risks. Instead, she argued that systemic absorption of fluoride is paramount.
“I want to make sure you understand that when we drink fluoridated water, we are allowing ourselves both a topical and a systemic source of fluoride. And both of these are important.” She said systemic ingestion is particularly important for young children.
Lewis said researchers arguing there is little to no benefit to swallowing fluoride are “biased.”
“What we’ve seen today is people cherry-picking studies and making conclusions without presenting us with the complete data that we need,” she added.
Those observations followed Taylor’s presentation, in which she explained that the National Toxicology Program and the JAMA publication presented an analysis of every available study, and they made all of their data publicly available.
Cole responded to Lewis, quoting her own Pediatrics in Review paper, in which she concluded the disadvantages of supplements were “substantial,” the benefits of fluoride were primarily topical and not systemic, that fluoride supplements cause more dental fluorosis and that their routine use is inconsistent with the way fluoride works — which is topically.
Michael Connett, lead attorney for plaintiffs in the lawsuit against the EPA, also read from Lewis’ paper during the meeting:
“The preponderance of strong research evidence supports the relative advantages of fluoride toothpaste over fluoride supplements, and this led Canada, England, Australia, New Zealand, and the European Union to recommend against regular use of fluoride supplements in favor of promoting fluoride toothpaste use in young children. The United States should do the same.
“FDA should ban these unapproved drugs from the market.”
Faced with her own research conclusion, Lewis conceded, “I personally think there’s a lot of disadvantages to supplements,” and said she would like to see the U.S. move to a model that promotes fluoride toothpaste.
Several public commenters raised concerns about the health impacts of fluoride. Then representatives from the ADA, American Academy of Pediatrics, other professional medical organizations, several dentists and dental hygienists submitted comments to the FDA advocating for the supplements to remain available.
They stated that the tablets have been proven “safe and effective.” None commented on the fact that they have never been studied.
Fluoride supplements carry ‘more risk than benefit,’ study says
The FDA facilitator referred to fluoride supplements by their official classification, “orally ingestible unapproved prescription drug products containing fluoride,” underscoring the fact that the drugs have never been subjected to an FDA approval process to determine if the benefits outweigh the risks.
The supplements were launched in the 1940s and later effectively grandfathered into the regulatory process. They never underwent the testing for safety and effectiveness typically required by FDA-regulated drugs, and the agency never granted them formal approval.
Before 1938, sodium fluoride had never been used in dentistry. Instead, it was commonly used as a roach and rodent poison. In 2016, FAN filed a citizens’ petition demanding the removal “of unapproved, unsafe, unnecessary, and ineffective sodium fluoride-containing” supplements from the market.
The petition cited a letter the FDA sent to Kirkman Labs, a fluoride supplement manufacturer, informing the company that it couldn’t sell its products because they were new, unapproved drugs not generally recognized as “safe and effective” to prevent dental decay.
The agency concluded that fluoride tablets didn’t meet the “generally recognized as safe” classification.
During public comments on Wednesday, Jay Sanders, FAN education & outreach director, cited a review in the Journal of Public Health Dentistry that found fluoride supplements “when ingested for a preeruptive effect by infants and young children in the United States, carry more risk than benefit.”
Sanders also noted that the CDC and the National Resource Council have both concluded that fluoride predominantly works topically, not systemically.
“A non-FDA approved drug with poor efficacy and with the potential to permanently damage the brain and disrupt the endocrine system should not be dispensed to children in the United States,” he said.
Tidmarsh underscored that understanding the risk-benefit analysis was key. “We’re not talking about taking a drug off the market that was already FDA-approved. In that context, we need to make sure there is a rigorous analysis.”
He added that makers of these drugs could always do real safety and efficacy testing on the products and submit them to the FDA for approval. “If we decide to take sodium fluoride supplements off the market, there’s nothing that would prevent a group from doing the rigorous studies and bringing it back to the FDA.”
This article was originally published by The Defender — Children’s Health Defense’s News & Views Website under Creative Commons license CC BY-NC-ND 4.0. Please consider subscribing to The Defender or donating to Children’s Health Defense.
FDA stalls decision on petition to suspend mRNA injections, citing ‘other priorities’
US regulator quietly delays action despite evidence of regulatory failure, DNA contamination, and a surge in cancers among young people.
By Maryanne Demasi, PhD | July 19, 2025
The U.S. Food and Drug Administration (FDA) has delayed its response to a formal petition demanding the suspension of the mRNA Covid-19 injections, citing “the existence of other FDA priorities.”
In a letter dated 17 July 2025, Dr Vinay Prasad—recently appointed Director of the FDA’s Center for Biologics Evaluation and Research (CBER)—acknowledged that the agency had “not yet reached resolution of the issues raised” in the petition.
Filed on 20 January 2025, the petition alleges that Pfizer’s Comirnaty and Moderna’s Spikevax were “unlawfully approved” in violation of federal regulatory requirements.
It calls for an immediate halt to the injections, independent testing of retained vials, and a full investigation into the approval process.
Fatal flaws in licensing mRNA products
Submitted by lawyer Katie Ashby-Koppens of PJ O’Brien & Associates, and spearheaded by former barrister Julian Gillespie, the petition argues that the mRNA injections were misclassified from the outset.
Although the products meet the FDA’s own definition of gene therapy, they were not regulated as such—sidestepping the heightened oversight normally required for gene-based interventions.
Under U.S. law, gene therapies must undergo ‘Environmental Assessments,’ be reviewed by specialised advisory committees, and face a more rigorous public transparency process.
But by labelling the mRNA injections as conventional ‘vaccines,’ regulators were able to fast-track their approval through a separate, less stringent pathway—bypassing critical safeguards.
The petition also raises alarm over synthetic DNA fragments found in the final products. Independent testing by multiple laboratories—including the FDA’s own facility—revealed DNA contamination far exceeding the safety limits.
Because the DNA is encapsulated in lipid nanoparticles, it can bypass normal immune defences, enter human cells, and in some cases integrate into the genome. The potential consequences, the petition warns, include genomic instability, cancer, and heritable genetic damage.
One of the most serious findings is the presence of SV40 promoter sequences in Pfizer’s injection—elements known to interfere with tumour-suppressing pathways such as p53.
The petition accuses Pfizer of withholding this information from the FDA in breach of disclosure laws.
Interim letter, no timeline
Under federal law, the FDA was required to respond to the petition within 180 days.
Just before the deadline, it issued a standard interim letter—acknowledging the petitioners’ main concerns but offering no timeline for a final decision.
Nor did the agency indicate that any investigation had begun. “We will respond to your petition as soon as we have reached a decision on your request,” wrote Prasad.
The agency’s delay is not uncommon—but critics say it reflects a deeper reluctance to confront the scientific and regulatory implications head-on.
Fully addressing the petition would require a sweeping and uncomfortable re-evaluation of how mRNA technologies were developed, approved, and marketed under the guise of conventional ‘vaccines.’
If the products were unlawfully licensed—mislabelled as vaccines to circumvent gene therapy regulations—the fallout would be unprecedented.
The admission alone could expose governments to extraordinary legal and financial liability—including product withdrawals, class actions, long-term health monitoring, injury compensation, and potential criminal investigations.
Petitioners speak out
Gillespie said the FDA is caught “between a rock and a hard place”—but that doesn’t excuse inaction. He believes the recent surge in cancers among young people demands urgent scrutiny.
“There’s been a tremendous and continuing rise in cancers across the United States commensurate with the rollout of these products,” he said. “Government officials have seen the data… and are refusing to address the elephant in the room.”
Analysis by Ethical Skeptic shows young cancers are up by 44%
Dr Jessica Rose, a computational biologist and co-author of the petition, said the public was never given accurate information about the nature of the products.
“The public was not told what they were being injected with,” she said. “And still to this day, they are not.”
She described the failure to distinguish gene-based therapies from traditional vaccines as “an existential crisis,” warning that “more and more people—including children and infants—are being exposed to the harms of foreign DNA.”
Dr David Speicher, a virologist and co-signatory on the petition, said the FDA’s letter amounts to bureaucratic minimisation.
“The number of vaccine-injured people continues to grow, and we do not all know the long-term harms caused by these genetic products,” he said. “Yet the FDA states that ‘other priorities’ are more important.”
He called for “an independent scientific team to examine the regulatory process, as well as to provide funding to researchers to explore biological mechanisms such as genomic integration.”
Pharmacy consultant and petitioner Maria Gutschi said the mRNA products represent a new therapeutic category “with no previous knowledge to leverage in assessing safety and efficacy.”
She argued that, given the novelty and risks, “the bar to suspend and/or mandate ‘black box’ warnings must be higher than for any previous therapeutic agent.” Gutschi urged the FDA to treat this as “THE priority” going forward.
A tale of two gene therapies
Critics say the FDA’s handling of mRNA harms stands in stark contrast to its swift response to safety concerns involving other gene therapies.
Yesterday, the agency announced a halt to clinical trials for Sarepta Therapeutics’ investigational gene therapy after the company reported another patient death—bringing the total to three deaths across two separate gene therapy products.
The treatment, developed for limb girdle muscular dystrophy, prompted immediate regulatory action.
“Today, we’ve shown that this FDA takes swift action when patient safety is at risk,” said FDA Commissioner Marty Makary, declaring the agency is “not afraid to take immediate action when a serious safety signal emerges.”
In contrast, the FDA has remained inert on mRNA injections—which also deliver genetic material into human cells but were classified as “vaccines”—despite thousands of reported deaths and serious adverse events following administration.
According to the petitioners, the public was led to believe they were receiving a conventional vaccine—when in fact, they were being administered gene therapy.
By failing to recognise and regulate the products accordingly, the FDA violated public trust—bypassing transparency laws, concealing critical risks, and depriving individuals of the opportunity to make informed medical decisions.
Next steps
People don’t want agencies to stall. They don’t want bureaucratic evasions. They want answers—and they want accountability.
The FDA’s next move won’t simply test regulatory process.
It will test courage—whether anyone inside the system is willing to confront the fallout in what may be the most consequential medical misclassification in modern history.
MenQuadfi Approval and the Pyramid Scheme of Vaccine Safety
This is how the game is played
Injecting Freedom by Aaron Siri | June 15, 2025
Recently, FDA shamefully approved Sanofi’s MenQuadfi (a meningococcal vaccine) to be injected into infants 6 weeks to 2 years old based on a trial that compared it to Menveo (another meningococcal vaccine). In the trial, 5.3% of infants receiving MenQuadfi and 3.6% of infants receiving Menveo had a serious adverse reaction (which means something very serious, see the FDA definition). But because these rates were “similar,” this product was deemed “safe” by FDA—because it assumes Menveo is also “safe.”
But Menveo was licensed based on a trial in which Menactra (among other vaccines) was used as a control; and Menactra was licensed based on a trial in which Menomune was used as a control; and Menomune was not licensed based on a proper placebo-controlled trial either. In fact—and this is mind-twisting—the package insert for Menomune lists the clinical trial for Menactra (in which Menomune itself was used as the control) as the basis for its safety. I couldn’t even dream of making this stuff up.
This provides a good example of the vaccine safety pyramid scheme: Menomune was licensed without a proper placebo-controlled trial and was then used as the control to license Menactra; Menactra is then used as the control to license Menveo; and then Menveo is used as the control to license MenQuadfi. And then we get a trial with 5.3% and 3.6% of infants suffering serious adverse reactions and FDA grants licensure.
What makes this even more troubling is that because FDA officials must know these numbers are highly concerning, they have Sanofi conduct a case-by-case review of each serious adverse event. If FDA were confident the control was safe, it would just rely on a statistical comparison between the vaccine being evaluated and the control. But since FDA officials must know Menveo’s safety is unknown (because it was licensed in a trial with a defective control), they ask Sanofi (the company seeking to get approval and profit from this product) to explain away each serious adverse event. And the Sanofi-paid researchers do exactly that in their write-ups to FDA about each serious adverse event.
And when FDA gets these write-ups explaining away each serious adverse event as “unrelated” to MenQuadfi, what does FDA do? The FDA officials reviewing them dutifully agree. What else would they do? Admit that Menveo, used as the control, and which they licensed based on nonsense data, has been harming children? That it is causing 3.6% of children—or even a fraction of that—to have a serious adverse event? If FDA officials do that, the house of cards would start to collapse. It would become clear Menveo wasn’t properly licensed (which it wasn’t), and that Menactra wasn’t properly licensed (which it wasn’t), and the same for Menomune.
FDA’s conflict and bias are dangerous. Letting Sanofi decide if its own product caused harm is beyond dangerous. This entire pyramid scheme, without a valid baseline of safety permitting a statistical comparison, requires injecting a new layer of biased assumptions with each additional licensure.
At this point, the safety of these products is based on dogma and assumptions. FDA has its reputation and any remaining trust to lose—and pharma its billions in profits—if they actually evaluated these products using a true safety comparator. It would reveal the true safety profile of these products (which the reliable data shows will likely be terrifying). Of course, the children whose injuries could be averted by conducting actual safety trials would benefit, but they are not really part of the equation.
References:
FDA branded ‘shameful’ over infant meningococcal vaccine approval
By Maryanne Demasi, PhD | June 8, 2025
“It’s shameful,” said attorney Aaron Siri of Siri & Glimstad LLP, criticising the FDA’s decision to expand use of the meningococcal vaccine MenQuadfi to infants as young as six weeks.
Previously licensed for children over two, the vaccine is now approved for babies aged 6 weeks to 23 months, based on trials in which infants received up to four doses.
Siri, who has represented families affected by vaccine injury, says the move reflects a broader pattern of weak oversight—where flawed trial designs and circular assumptions are used to justify approvals despite serious safety concerns.
Serious adverse events in infants
According to the FDA’s own summary, 5.3% of infants who received MenQuadfi in clinical trials experienced at least one serious adverse event (SAE)—defined as any medical occurrence resulting in death, hospitalisation or disability.
That figure reflects SAEs reported from the first dose through six months after the final dose.
That’s roughly one in every 20 children.
In the comparator group, 3.6% of infants who received Menveo also experienced a serious adverse event during the same period.
Instead of raising concern, the FDA took comfort in the similarity.
The agency concluded that because the rates of serious reactions were “comparable,” the expanded use of the vaccine in infants could be considered safe.
But that logic, Siri argues, is dangerously circular. “Because these rates were ‘similar,’ this product was deemed ‘safe’ by FDA because it assumes Menveo is ‘safe.’”
In the end, Sanofi, the company selling MenQuadfi, chalked up only two cases as possibly related to vaccine—both febrile seizures after the 4th dose, given in combination with MMR, varicella, and PCV13 at 12 months of age.
A placebo problem
The issue, Siri says, lies in the chain of assumptions.
Menveo wasn’t tested against a true saline placebo. It was compared to Menactra—another meningococcal vaccine. Menactra, in turn, was tested against Menomune.
But Menomune—now discontinued—was never tested against a placebo either.
The final twist?
In a bizarre loop of regulatory logic, the package insert for Menomune cites the clinical trial for Menactra—where Menomune was the comparator—as part of its own safety justification.
“I couldn’t even dream of making this stuff up,” Siri said.
What emerges is a kind of regulatory ouroboros—a snake swallowing its own tail—where each new product is built on the presumed safety of the last, and none are ever measured against a neutral baseline.
The vaccine safety pyramid
“This provides a good example of the vaccine safety pyramid scheme,” said Siri.
“Menomune was licensed without a proper placebo-controlled trial and was then used as the control to license Menactra; Menactra is then used as the control to license Menveo; and then Menveo is used as the control to license MenQuadfi,” he added.
Each step is built on the presumed safety of the one before it—but without any solid foundation. No inert comparator. Just a chain of assumptions.
“Hence, we get a trial with 5.3% and 3.6% of infants suffering serious adverse reactions and no one bats an eye—they grant licensure,” Siri said.
How did the FDA allow this?
Under current FDA guidelines, vaccine manufacturers are not required to use a placebo—an inert substance such as a saline injection—in pre-licensure trials. Instead, “active comparators” such as other vaccines are commonly used.
This practice speeds up approval, especially when companies argue the control group must be “protected” from the disease being targeted.
But critics say it undermines transparency and obscures harms.
If both the test vaccine and the comparator cause adverse events, the trial may appear to show no safety signal—even if the absolute rate of harm is high.
In this case, the FDA accepted the MenQuadfi trial design without requiring a true placebo group—despite the high rate of reported serious events.
Who pays the price?
Since MenQuadfi is already on the CDC schedule for older children, the pharma companies profiting from this product already have liability protection under U.S. vaccine injury laws.
“FDA and pharma have nothing to lose here,” Siri said.
“We, as taxpayers, will pay for all of the harms suffered and, worst of all, the children who are injected and harmed and their families will really pay for the harms,” he added.
It’s not the first time critics have accused the FDA of playing fast and loose with vaccine safety data, especially in the wake of Covid-19.
But this case, Siri argues, is a textbook example of how safety assessments are gamed when it comes to childhood vaccine.
Restoring integrity in vaccine testing
At the heart of Siri’s warning is a plea for scientific integrity.
Without true placebo-controlled trials, he argues, there’s no meaningful way to detect harms—just one untested product being measured against another.
“This isn’t science. It’s a shell game,” he said.
As MenQuadfi rolls out to younger babies, public health officials will no doubt emphasise its potential to prevent a rare but deadly disease.
But advocates like Siri want parents and legislators to understand what’s beneath the product label—not just a vaccine, but a regulatory system that is collapsing under the weight of its own shortcuts.
Featured Video

10 MINUTES COVID MADNESS
or go to
Aletho News Archives – Video-Images
From the Archives
Why Hitler Declared War on the United States
By John Wear | Inconvenient History | 2017-08-31
Establishment historians state that Adolf Hitler made a mistake when he declared war on the United States. For example, British historian Andrew Roberts wrote:[1]
“It seems an unimaginably stupid thing to have done in retrospect, a suicidally hubristic act less than six months after attacking the Soviet Union. America was an uninvadable land mass of gigantic productive capacity and her intervention in 1917-18 had sealed Germany’s fate in the Great War.”
Historian Martin Gilbert wrote in regard to Germany’s declaration of war on the United States:[2]
“It was perhaps the greatest error, and certainly the single most decisive act, of the Second World War.”
In this article I will explain why Hitler was forced to declare war on the United States. … continue
Blog Roll
 Aletho News
Aletho News- Netanyahu will reject US demands for withdrawal from Lebanon, Gaza, Syria if asked: Report
- Russia warns Ukraine expanding ‘terrorist activities’ beyond Europe
- US use of Italian military bases in the conflict with Iran shows lack of autonomy
- The synthesis of regional conflicts into WWIII has begun
- Is the Department of War Covering Up the True Extent of US Casualties?
- Is Peter Mandelson a Russian or a Zionist asset?
- German arms exports to Israel surge sharply amid Gaza genocide
- Massie says the US has been ‘significantly weakened’ by war on Iran
- Zelensky: North Korea, Iran and China Backing Russia’s War in Ukraine
- Saudi oil loading volumes drop 40 percent at Red Sea port amid Yemeni blockade
- If Americans Knew
- ‘Hell on Earth’: Roger Waters on Palestine, Apartheid and the Cost of Speaking Out
- ISRAELI MEDIA: Israelis Fight Over Framing of West Bank Attacks. But Settler Violence Is an Israeli Strategy (2 articles)
- Attacks on Mamdani Prove Israel’s Supporters Can’t Even Tolerate Criticism of Netanyahu
- Integrated Israeli Policies Fast‑Track Israeli Annexation of Palestinian Land
- ICC Chief Prosecutor Karim Khan dared to hold Israel to account – that’s why he was brought down
- Why did Bari Weiss fire Cecilia Vargas?
- Rescue teams recover remains of 99 Palestinians from one home in Gaza – Daily Update
- Patrick Bet-David sells wealth building, but the real product is Zionist shilling
- The Last-Minute Million-Dollar Ad Spend Against Will Lawrence Is Coming From a Major Pro-Israel Group
- Five decades of Israeli covert strategy to push US, Iran into confrontation
- No Tricks Zone
- ECMWF Models Throwing Cold Water On Extreme Germany Heat Wave Forecast Next Week
- Greenland’s Ice Sheet Was Supposed To Be Rapidly Melting Away. It Hasn’t Been.
- Global Temperature Trend Has Cooled Over The Past 6500 Years, Scientists Have Found
- Wind Energy Means Going Back To The Middle Ages, Says German Professor Horst-Joachim Lüdecke
- New Study: A 40-Fold Increase In Earth’s Main Greenhouse Gas Contributes To Cooling The Ocean
- New Study Highlights The ‘Dominant Role’ Of Aerosol/Cloud Interactions In Shaping Climate
- Munich’s First-Ever Green Party Mayor Declares First Ever City Water Use Restrictions… Fines Up to 50,000 €!
- Experimental Lab Research: The Climate Sensitivity To A 400-Fold Increase In CO2 Is 0.1°C
- Fatal Snobbery: In France, It’s Better To Die From A Heatwave Than To Do As Americans
- New Study: NASA’s Models Wildly Underestimate The Capacity Of Clouds To Alter Solar Radiation

{kind=link}