U.S. Government ‘Saddled’ With COVID Vaccine Injury ‘Mess’ — While Vaccine Makers Avoid Liability
By Michael Nevradakis, Ph.D. | The Defender | July 18, 2024
As early as January 2022, National Institutes of Health (NIH) researchers were aware of at least 850 peer-reviewed case reports and/or research articles about COVID-19 vaccine reactions, according to emails obtained by Children’s Health Defense (CHD).
In one email (name and agency redacted), NIH researchers were told the federal government was “saddled” with the “mess” of dealing with those injured by the COVID-19 vaccines, due to the liability shield enjoyed by vaccine manufacturers.
The emails, part of a 309-page batch of documents released to CHD on June 21, originated from a U.S. Food and Drug Administration (FDA) request to NIH researchers for input on a report highlighting several injuries common among people who received the vaccines.
CHD requested the documents via a Freedom of Information Act (FOIA) request to the NIH in November 2022. When the NIH hadn’t responded by April 2023, CHD sued the agency.
In an October 2023 settlement, the NIH agreed to produce up to 7,500 pages of documents at a rate of 300 pages per month.
The batch of documents released in June — which include emails to Peter Marks, M.D., Ph.D., director of the FDA’s Center for Biologics Evaluation and Research — revealed that by fall 2021, key NIH researchers were aware of scientific studies on serious adverse events, including persistent neurological symptoms, following COVID-19 vaccines.
As with prior releases of the NIH documents, June’s tranche also included several emails from vaccine-injured individuals to NIH researchers, seeking help for their symptoms — with one person asking, “Why aren’t you studying vaccine injuries?”
‘Tinnitus … was a freight train in my head for the first four months’
On Jan. 10, 2022, NIH researcher Dr. Avindra Nath was forwarded an email from someone whose name is redacted, with the subject line: “Followup [sic] Jan 4th Meeting” (pages 281-289).
The original email, dated Jan. 9, 2022, was sent to FDA officials including Marks and Dr. Janet Woodcock, principal deputy commissioner of food and drugs, who apparently participated in a meeting on this topic on Jan. 4, 2022.
The Jan. 9, 2022 email included a list of “persistent symptoms following the Covid vaccines” and the names of researchers who were studying these conditions, which included dysautonomia, neuropathy, tinnitus, multisystem inflammatory syndrome (MIS), myocarditis, blood clots and parasthesias.
The email was accompanied by a spreadsheet listing approximately 850 “peer-reviewed case reports/research articles about Covid vaccine reactions.”
Regarding dysautonomia — a nervous system disorder that disrupts automatic bodily functions — the email stated that the condition is “grossly under diagnosed” and “is not diagnosed in ERs or ICUs” but in “autonomic specialty labs.”
The email noted that such labs are less likely than hospitals to file reports with the Vaccine Adverse Event Reporting System (VAERS) and added that there “likely are issues with identifying this syndrome if only looking through VAERS or similarly reported databases.”
As a result, the email suggested “it would be reasonable to approach autonomic specialists / long covid specialists about their observations.”
A 2011 Harvard study found that less than 1% of all adverse events are reported to VAERS.
The Jan. 9, 2022, email also noted unusual trends regarding diagnoses of neuropathy — a set of neurological symptoms that includes numbness and tingling in the hands or feet, and a burning, stabbing or shooting pain in affected areas.
According to the email, “Historically, neuropathy presents in the predominantly male population aged 59+. However as discussed previous [sic], neuropathy in our case is predominantly female, aged 29-40.”
As with dysautonomia, the email noted that neuropathy is “likely to be inadequately reported through the VAERS and BEST [Biologics Effectiveness and Safety] systems because of the circumstances previously mentioned for dysautonomia.”
The Jan. 9, 2022 email also acknowledged that tinnitus was a common post-vaccination injury, noting, “Our findings are that this is not just J&J [the Johnson & Johnson, or Janssen, COVID-19 vaccine] … not by a long shot.”
According to the email, “This symptom is more proportionate to the general neuro symptoms by brand as previously reported in our patient led survey of 500 participants.”
The email’s author also noted that, “in my case yes, I have tinnitus now and it was a freight train in my head for the first four months.”
‘Is it reasonable to dismiss … 20 new symptoms … in a single person post vaccine?’
According to the email, myocarditis and blood clots were already “acknowledged by the FDA and CDC” (Centers for Disease Control and Prevention).
“Every person in our groups that have one of these two conditions, also have accompanying neuro issues like those of us who are not currently acknowledged by the FDA and CDC,” the email said.
The conditions included postural orthostatic tachycardia syndrome (POTS), “brain fog/memory loss, and inflammation (MCAS)” — mast cell activation syndrome.
“Even the perfectly healthy very fit young males with the lasting myocarditis are struggling with the POTS and inflammation/brain fog/memory loss. Makes me suspect that somehow these all are a result of the same mechanism of action,” the email stated.
The Jan. 9, 2022, email also acknowledged parasthesia — a condition that causes a burning, prickling sensation — and MIS, a condition in which numerous organs become inflamed, as concerns.
The email openly questioned why more wasn’t being done to connect these conditions in the vaccinated, to the COVID-19 vaccines themselves, noting that vaccinated people were frequently demonstrating multiple rare symptoms:
“While we understand that correlation does not equal causation, we also find a strong correlation with the change in our blood that mirrors long-haul, and symptomology that mirrors long-haul.
“Because of this, I have to ask what is the process by which Covid PASC [post-acute sequelae of SARS-CoV-2 infection, or long COVID] symptoms have been so readily tied back to Covid, whereas the same symptoms due to the Covid vaccines have not?
“Also, while it may be coincidental to have one or maybe two strange symptoms pop up, is it reasonable to dismiss 10, 15, 20 new symptoms that occur in a single person post vaccine.”
‘Insanely challenging for these people suffering … to walk this path alone’
In the Jan. 10, 2022, email to Nath an NIH researcher wrote, “The FDA has asked once again for us to provide any input from those who have experience with this disease. Very prompt responses and more active engagement on their part lead me to believe they will now examine these problems with some effort.”
The author also asked Nath if he knew researchers “who could fill in the gaps” and asked him if he would “kindly be willing to discuss with Peter Marks?”
“The gov has conveniently absolved the drug companies of any liability, and the federal government is now saddled with the responsibility of figuring out this mess,” the email continued. “I am happy to orchestrate a meeting of the minds with NDR [non-disclosure] agreements if that would get the discussion started in a way that is similar to how previous new diseases have been investigated.”
The email also noted talks with public health officials in Germany and France.
“It has been insanely challenging for these people suffering to have to walk this path alone. They grow more and more desperate by the day. Knowing there is someone, somewhere looking into this makes a big difference for these people to just hang on.”
Even though public health agencies were aware of this information and were discussing vaccine injuries in early 2022, official government advice to the public continued to claim the COVID-19 vaccines were “safe and effective,” including statements by Dr. Anthony Fauci in November 2022.
And in testimony before Congress in February, Marks dismissed the COVID-19 vaccine injury reports filed with VAERS, stating that numerous false reports are submitted to the database — a claim some experts have disputed.
As of today, the CDC continues to recommend the COVID-19 vaccines “for everyone ages 6 months and older, including people who are pregnant, breastfeeding, or might become pregnant in the future.”
NIH researchers aware of vaccine injury studies in fall of 2021
The June 2024 tranche of NIH documents also revealed that, at least as early as fall 2021, researchers with the agency were aware of scientific studies and surveys highlighting serious adverse events following COVID-19 vaccination.
In a Sept. 2, 2021, email (pages 109-121), Farinaz Safavi, M.D., Ph.D., of the NIH Division of Neuroimmunology and Neurovirology was sent the results of the “Covid Vaccine Persistent Symptoms Survey” conducted by React19, a group advocating on behalf of COVID-19 vaccine injury victims.
The version of the survey included in the email was accurate as of Aug. 31, 2021, and contained the results of 382 questionnaires submitted by people “suffering persistent neurological symptoms after receiving the Sars-CoV2 Vaccine in the United States.”
According to those results, 71% of respondents said they had no preexisting health conditions prior to the symptoms they developed following their COVID-19 vaccination, and 94% said they had never previously experienced a reaction to other vaccines.
The most commonly reported symptoms included paresthesia, tinnitus, heart palpitations, tachycardia, chest pain, visual disturbance or loss, muscle twitching, joint pain, muscle aches, brain fog, fatigue and anxiety attacks.
Almost all respondents said these symptoms began less than two weeks following vaccination.
In a Nov. 15, 2021, email (pages 300-305), Nath was sent a scientific paper, “Neurological side effects of SARS-CoV-2 vaccinations,” authored by Austrian researcher Josef Finsterer, M.D., Ph.D.
According to this paper, “The most frequent neurological side effects of SARS-CoV-2 vaccines are headache,” Guillain-Barré syndrome, venous sinus thrombosis and transverse myelitis.
“Safety concerns against SARS-CoV-2 vaccines are backed by an increasing number of studies reporting neurological side effects. … Healthcare professionals, particularly neurologists involved in the management of patients having undergone SARS-CoV-2 vaccinations, should be aware of these side effects and should stay vigilant to recognize them early and treat them adequately,” the paper concluded.
Nath received a review copy of this paper, which has since been published in Acta Neurologica Scandinavica.
And in a May 17, 2021, email (pages 292-299), Nath was sent a preprint of “Sudden Onset of Myelitis after COVID-19 Vaccination: An Under-Recognized Severe Rare Adverse Event,” co-authored by William E. Fitzsimmons, doctor of pharmacy, and Dr. Christopher S. Nance.
According to the preprint, “Myelitis has been reported as a complication of COVID-19 infection. However, it has rarely been reported as a complication of COVID-19 vaccination.”
The paper focused on the example of one of Fitzsimmons’ patients, a 63-year-old previously healthy male who developed myelitis after his second dose of the Moderna COVID-19 vaccine — and treatment that was effective in his case.
Other emails apparently sent by Fitzsimmons highlighted the injuries and the progression of treatment of this 63-year-old man (pages 145-150).
‘A blood clot as a cause of your paralysis would make the most sense’
In an email chain to Nath beginning Sept. 20, 2021, (pages 228-233) with the subject “Paralyzed after J&J Covid Vaccine,” the author (whose name is redacted) said that less than 24 hours following vaccination, the patient “lost bladder control.” He later developed a blood clot and erectile dysfunction, before becoming paralyzed.
In a response that day, Nath told the patient, “The temporal association of the symptoms with the vaccine does make is [sic] suspect, but I do not know of any way how to sort it out.”
In a follow-up email that day, Nath said, “A blood clot as a cause of your paralysis would make the most sense, however, proving cause and effect related to the vaccine in a single patient is virtually impossible.”
In a Dec. 13, 2021, email to Nath (pages 234-236), another vaccine injury victim, who “was healthy prior to vaccination,” described injuries following both doses of the Pfizer-BioNTech COVID-19 vaccine, including paresthesia, tachycardia, severe tinnitus, intractable insomnia and “POTs-like symptoms.”
“I have been diligent and determined in seeking care near and far, but have continued to face skepticism, half-interest, and an inability to know how best to treat,” this person wrote.
And in a series of emails beginning Jan. 24, 2022, (pages 246-247), a “woman who was completely healthy before taking the Pfizer vaccines” told Nath about a series of neurological symptoms and inflammation she experienced following her second dose, in addition to symptoms like tinnitus, insomnia and brain fog.
“Why isn’t the NIH doing research on this?” she asked in a follow-up email on Jan. 25, 2022.
This article was originally published by The Defender — Children’s Health Defense’s News & Views Website under Creative Commons license CC BY-NC-ND 4.0. Please consider subscribing to The Defender or donating to Children’s Health Defense.
PCR Testing for Bird Flu ‘Will Only Serve to Raise False Case Count’ Critics Say
By Brenda Baletti, Ph.D. | The Defender | June 6, 2024
Dr. Deborah Birx, the Trump administration’s coronavirus response coordinator, told CNN’s Kasie Hunt the U.S. is making the “same mistakes” with bird flu that it made with COVID-19, which she said spread because there wasn’t enough testing for asymptomatic infection.
Birx is now calling for every cow to be tested for bird flu weekly and for regular pooled tests for dairy workers. She also said it’s likely that undetected cases are circulating in humans.
“We have the technology,” Birx said. “The great thing about America is we’re incredibly innovative and we have the ability to have these breakthroughs.”
The technology Birx referenced is polymerase chain reaction or PCR testing — the same diagnostic tool that came under fire during the COVID-19 pandemic for producing inaccurate results, including false positives.
Speaking out on X (formerly Twitter), critics like Simon Goddek, Ph.D., pushed back, accusing Birx of “deliberately using the same strategy to fabricate another fake health emergency.”
On Wednesday, the day after Birx’s interview, JAMA published its own article advocating for more widespread bird flu testing.
“No animal or public health expert thinks that we are doing enough surveillance,” Keith Poulsen, DVM, Ph.D., director of the Wisconsin Veterinary Diagnostic Laboratory at the University of Wisconsin-Madison, told JAMA.
Andrew Pekosz, Ph.D., from the Johns Hopkins Bloomberg School of Public Health, told JAMA that more testing should be conducted to find asymptomatic and mild infections. Workers at infected farms should be tested twice weekly, he said, and cows should be tested once a week.
Inventor: PCR test never intended for use as diagnostic tool
PCR testing works by starting with tiny fragments of DNA or RNA called nucleotides and replicating them until they become large enough to identify. The nucleotides are replicated in cycles, and each cycle doubles the amount of genetic material in the sample. The number of cycles required to create an identifiable sample is the “cycle threshold” (Ct).
PCR tests became a household name during the COVID-19 pandemic because they were treated as the “gold standard” for identifying positive cases, especially among asymptomatic people.
However, as early as December 2020, the World Health Organization (WHO) warned that using a high-cycle threshold would lead to false-positive results. The agency encouraged healthcare providers to consider the test in concert with other factors — namely the presence of symptoms — when diagnosing patients.
The WHO also cautioned those using the tests to read the instructions carefully to determine whether the cycle threshold ought to be changed to account for any background noise that could lead to a high-cycle threshold being mistaken for a false positive.
“When specimens return a high Ct value,” the press release said, “it means that many cycles were required to detect virus. In some circumstances, the distinction between background noise and actual presence of the target virus is difficult to ascertain.”
Kary Mullis, who won the Nobel Prize for inventing the PCR test, said it was inappropriate to use the test as a diagnostic tool to detect a viral infection.
Even Dr. Anthony Fauci admitted during the pandemic that a high cycle — which was used often — detected only “dead nucleotides,” not a viral infection.
The U.S. Department of Agriculture (USDA) did not immediately respond to The Defender’s inquiry about which cycle thresholds are used to test animals for bird flu.
Mass testing ‘will only serve to raise a false case count’
As of Tuesday, the latest circulating bird flu virus has reportedly infected 81 herds of dairy cattle in nine states and poultry farms in 48 states. The virus can be fatal for poultry but does not generally cause serious illness in cattle.
Bird flu is rare among humans. The Centers for Disease Control and Prevention (CDC) maintains it poses only a low risk to public health.
In the latest wave of bird flu, only three people in the U.S. have tested positive for the virus after close exposure to an infected cow. All three experienced mild symptoms — two experienced eye irritation and one also had a cough and sore throat. All recovered without incident.
The WHO reported Wednesday that a resident of Mexico died from a bird flu infection, but WHO officials also maintain the virus’ threat to the general population is low.
Bird flu cannot be transmitted among humans, but that hasn’t stopped health officials such as the WHO’s Chief Scientist Jeremy Farrar and U.S. Food and Drug Administration Commissioner Robert Califf from publicly stoking fears that the virus could suddenly mutate, become more infectious and transmissible among humans, and cause a pandemic.
Mainstream media outlets like Scientific American warned that the bird flu isn’t a pandemic “yet,” but it could evolve to become one if people do things like continue to drink raw milk. And The New York Times warned yesterday that the virus “may not be done” adapting.
The CDC reported on Tuesday that it monitors genetic changes in the virus and “few genetic changes of public health concern have been identified.”
Nevertheless, the U.S. government is building up its national stockpile of existing vaccines produced by CSL Seqirus and is nearing contracts with Moderna and possibly Pfizer to fund the development of an mRNA vaccine for the virus.
On Tuesday, Finland announced it will begin offering the vaccine to selected groups of people.
Other public health experts have dismissed the alarmism as “overblown,” with some suggesting the “fearmongering” is motivated by profit.
Dr. David Bell, a public health physician and biotech consultant, told The Defender last month the bird flu scare was “farcical.”
“We did not have a bad outbreak for over a century, and there is every likelihood that we won’t again,” Bell said. “We are using technology to pretend that new threats are occurring because we can now detect them.”
Cardiologist Dr. Peter McCullough said last month that mass testing of healthy animals — as Birx is suggesting — will only serve to “raise a false case count.”
Feds using PCR testing on animals, wastewater, farmworkers, meat and milk
The federal government last month announced a new round of funding to reduce the impact of bird flu. The plan appropriated $93 million for the CDC to do virus genomic sequencing, increase monitoring of farmworkers, and improve and expand testing on a national scale for bird flu in animals, wastewater, farmworkers and meat.
The FDA also appropriated an additional $8 million to surveil and test the commercial milk supply.
Lactating dairy cows must be tested for bird flu before they can cross state lines, per an April 24 Federal Order issued by the USDA.
The USDA’s Animal and Plant Health Inspection Service (APHIS) also encourages farmers to voluntarily test cattle and herds with suspected infection, showing symptoms like reduced milk production or respiratory issues. APHIS covers the cost of the tests if conducted at an approved laboratory and if the farmers agree to have the tested cattle and premises tracked.
The approved laboratories conduct PCR tests for several different flu strains that could be bird flu markers, including “FluA matrix, H5 and optionally H5N1 2.3.4.4b,” to determine whether the cattle are infected.
All laboratories, whether or not on the USDA’s approved list, must report all positive influenza A test results to the USDA weekly by 5 p.m. on Mondays. Farms with positive cases are quarantined.
KFF Health News reported that additional testing of farmworkers would make it possible for researchers to “track infections.” The problem is that “people generally get tested when they seek treatment for illnesses,” but farmworkers don’t tend to go to doctors unless they are very ill.
Farmworkers have been actively monitored for symptoms since the first case was detected but not PCR-tested. In response, federal authorities announced in May they would pay farmworkers to get tested for the virus as part of a program that also offers incentives to farmers to allow their dairy herds to get tested.
Workers are paid $75 for giving the CDC a blood sample and nasal swab.
The federal money also goes to support new wastewater surveillance using PCR tests. The CDC’s National Wastewater Surveillance System, launched in 2020, collects and makes public viruses identified in facilities across the country.
That wastewater testing is done by organizations including WastewaterSCAN, an infectious disease monitoring program based at Stanford University, in partnership with Emory University and funded through philanthropy, including the Sergey Brin Family Foundation created by the founder of Google.
The organization has most recently detected bird flu in San Francisco, although it is unclear whether it comes “from animal waste, milk, people or a combination of sources, according to the Los Angeles Times. WastewaterSCAN on June 3 began publicly reporting H5 data from its 190 sampling sites across the country in its online dashboard.
Other private companies that do PCR-based wastewater surveillance, like Biobot Analytics, are funded by venture capital firms in addition to the CDC.
The USDA Food Safety and Inspection Service is also testing meat from condemned cows. Late last month the agency announced that 95 of 96 culled cows tested negative for viral particles and that none of their meat entered the food supply.
It also reported that ground beef from retail facilities in states with cows that have tested positive for bird flu was all PCR-negative for the virus.
The agency also experimented with inoculating meat with high levels of the virus and then cooking it and testing for the virus. The virus was not detected in the meat patties cooked to medium or well-done, and it was “substantially inactivated” in the rare patties.
The FDA also tested retail dairy products in 17 states. The agency noted that PCR-positive results “do not necessarily represent live virus that may be a risk to consumers,” so when they found PCR-positive samples, they further tested them through a process called “egg inoculation.”
The agency found many samples with positive PCR tests, but none tested positive for the live virus.
This article was originally published by The Defender — Children’s Health Defense’s News & Views Website under Creative Commons license CC BY-NC-ND 4.0. Please consider subscribing to The Defender or donating to Children’s Health Defense.
Brenda Baletti, Ph.D., is a senior reporter for The Defender. She wrote and taught about capitalism and politics for 10 years in the writing program at Duke University. She holds a Ph.D. in human geography from the University of North Carolina at Chapel Hill and a master’s from the University of Texas at Austin.
FDA Approves Moderna’s mRNA RSV Vaccine — With No Input From Independent Advisers
By Brenda Baletti, Ph.D. | The Defender | June 3, 2024
The U.S. Food and Drug Administration (FDA) last week approved Moderna’s mRNA respiratory syncytial virus (RSV) vaccine for adults age 60 and older.
The FDA approved the drug without input from the agency’s independent vaccine advisory committee, which typically makes recommendations about such drugs, because the FDA didn’t see any “concerns or controversial issues” that would make input necessary to the approval process, the agency said in its approval letter.
Moderna is running at least 11 clinical trials for its new mRNA RSV drug on several other demographic groups, including young children, adolescents and healthy adults.
The vaccine, marketed under the name mResvia, is Moderna’s second-ever FDA-approved drug. It uses the same mRNA platform as its COVID-19 Spikevax vaccine.
“The FDA approval of our second product, mRESVIA, builds on the strength and versatility of our mRNA platform,” said Moderna CEO Stéphane Bancel.
The Centers for Disease Control and Prevention (CDC) must recommend the drug before it can be used. The CDC’s advisory committee will discuss and vote on the vaccine at its meeting next month.
If approved, the vaccine will provide a second revenue stream for Moderna, whose first-quarter sales fell 91% compared with same-quarter sales in 2023.
The company said it expects to launch the vaccine in time for the 2024 fall vaccination season.
The FDA’s approval of Moderna’s mRNA RSV vaccine comes a year after the agency approved GSK’s Arexvy and Pfizer’s Abrysvo RSV vaccines for the same age group.
Abrysvo also is approved for pregnant women. Pfizer is seeking approval for its drug for adults ages 18 and older and is testing it on children and teens. GSK is seeking approval for Arexvy for people 50 and above and expects a response this month.
Bancel said Moderna’s shot has an advantage over the RSV vaccines now on the market because it comes in a pre-filled syringe, making it faster to administer and cutting the risk of administration errors.
Moderna said it hopes to capture part of what it predicts will be a $10 billion market for RSV vaccines, especially given its post-pandemic plummet in profits.
The company also announced last week that it is in talks with the U.S. government to fund late-stage trials of its mRNA bird flu vaccine.
Experts say efficacy exaggerated and safety concerns ignored
Moderna’s approval was based on findings from a Phase 3 study published in December 2023 of more than 35,000 adults across 22 countries that claimed the vaccine was 83.7% effective at preventing at least two symptoms of RSV, such as cough and fever, nearly four months post-vaccination.
Follow-up analysis by the FDA identified other cases and reduced the efficacy to 79%, the company said. That efficacy rate is in line with Arexvy, which currently dominates the RSV vaccine market, Reuters reported.
Cardiologist Dr. Peter McCullough wrote on his Substack that the efficacy claims were misleading.
“The absolute risk reduction for significant outcomes was far below 1%, meaning this product will not have a significant clinical impact,” he wrote.
Absolute risk reduction refers to the actual difference in risk between the treated group versus the control group.
Relative risk reduction, which is how the company presented its trial data, is a proportional measure of how much a treatment reduced the risk of a bad outcome relative to the control group. It tends to lead to overestimations of how effective a treatment is.
Data presented in February also showed Moderna’s shot has faster efficacy declines compared to the GSK and Pfizer shots, Reuters reported.
Moderna said there were no serious safety concerns identified in the trial. Adverse reactions reported in the clinical trial included injection-site pain (55.9%), fatigue (30.8%), headache (26.7%), muscle pain (25.6%), joint pain (21.7%), underarm swelling or tenderness (15.2%) and chills (11.6%).
There were also two cases of acute pericarditis, which occurred after 42 days. The investigator considered those cases to be unrelated to the shot.
There were no reported cases of Guillain-Barré syndrome, which both Pfizer and GSK identified in their clinical trials and has subsequently proven to be “more common than expected,” with the other RSV shots, according to the CDC.
However, McCullough said that for all mRNA shots, there are concerns about myocarditis, auto-immunity, genomic integration and oncogenicity. The rapid approval process does not allow the necessary time to identify a lot of these issues.
He wrote:
“Our great concern was that mRNA COVID-19 vaccines ushered in the context of an emergency would set a new precedent for more genetic vaccines that depart from all safety standards set forth previously by the US FDA. …
“[The approval] was done without the full dossier of safety information required for a routine approval including 2-3 years of observation for standard vaccines, and at least 5 to 15 years of observation for genetic transfer technology.”
Because the FDA’s vaccine advisory committee didn’t discuss the data, there was no publicly accessible discussion of the vaccine’s efficacy and risk or space for public comment, which typically happens at such meetings.
The committee held meetings before the approval of both Arexvy and Abrysvo.
The FDA did not immediately respond to The Defender’s inquiry about the lack of an advisory committee meeting or possible concerns with vaccine safety.
RSV is a common respiratory virus that usually causes mild cold-like symptoms, but in some cases can lead to hospitalization and death in infants and the elderly.
The number of people who get RSV is unknown because the virus is rarely diagnosed unless one comes to a hospital and is tested.
Dr. Meryl Nass, an internist, told The Defender that among older people typically only those who are already ill or have very severe immune deficiency could benefit from an RSV vaccine.
“That benefit,” she said, “must be weighed against all the harms, including those from the lipid nanoparticle as well as the mRNA and any DNA plasmids or other extraneous production materials.”
She said that mRNA vaccines are typically expensive and the amount of spending that would be necessary to save one life could take away from other essential health spending.
McCullough said, “Rare illnesses which are mild should not be the target for mass vaccination.”
“In the case of respiratory syncytial virus, the illness is so mild and easily treatable with albuterol and budesonide nebulizers, it is hard to make the case for mass vaccination with a novel mRNA platform,” he added.
Brian Hooker, Ph.D., chief scientific officer of Children’s Health Defense, told The Defender that approval of this vaccine “is an absolute disaster in the making.”
“The clinical trial was too short (average 112 days) to ascertain any long-term sequelae to the vaccine. Even with that, the rate of serious adverse events was 2.8% or 1 in 36 vaccine recipients,” he said. “We can only expect the actual degree of damage will be much worse.”
Moderna conducting testing on pregnant women and their infants
A search of the federal clinical trials database also revealed that Moderna is testing its mRNA RSV vaccine on pregnant women and their infants, despite concerns raised among this group with other RSV vaccines.
The ongoing Phase 2 trial in pregnant women will consist of 360 participants between 28 and 36 weeks of gestation at the time of vaccination. The trial is designed to determine dosing and potential adverse events associated with the vaccine.
GSK halted the development of its RSV vaccine for pregnant women when it found a safety signal for preterm births among vaccinated women. In that study, for every 54 infants born to women who received the vaccine, one additional preterm birth occurred.
Neonatal deaths — the death of an infant in the first 28 days of life — also were higher in the GSK vaccine group, occurring in 0.4% of the infants in the vaccine group (13 of 3,494) and 0.2% in the placebo group (3 of 1,739), which they also noted was not statistically significant.
The FDA approved Pfizer’s Abrysvo for pregnant women in August 2023.
Pfizer’s own clinical trial data for Abrysvo, which is very similar to GSK’s vaccine, also showed elevated rates of preterm birth among vaccinated women, but the higher rates were not statistically significant, Pfizer said.
Still, the FDA limited approval of the vaccine for women in weeks 32-36 of their pregnancy to reduce risk and mandated post-market follow-up studies for both preterm birth and eclampsia.
The agency also labeled preterm birth as a potential risk associated with the vaccine.
Some members of the FDA’s vaccine advisory committee said they had serious safety concerns based on the clinical trial data, and four members voted against approving the drug.
And a recent preprint study shows a statistically significant safety signal for preterm birth associated with Abrysvo.
Clinical trials for the COVID-19 mRNA vaccines did not include pregnant women.
However, subsequent research found the mRNA administered to lactating mothers spread systemically from the injection site to breast milk. Other post-marketing studies of the COVID-19 vaccine found mRNA in umbilical cord blood and in the placenta.
Moderna also has several other active clinical trials for the drug, including among people who are not at risk from RSV-related illness, including children and adults, children ages 2-18, and healthy adults, among others.
It is also testing the drug among children ages 5-24 months.
This article was originally published by The Defender — Children’s Health Defense’s News & Views Website under Creative Commons license CC BY-NC-ND 4.0. Please consider subscribing to The Defender or donating to Children’s Health Defense.
Informed Dissent
Medical Dissidents, Agency Capture, and Dr. Mary Talley Bowden’s Battle with the FDA over Ivermectin

BY M.C. ARMSTRONG | HONEST MEDIA | APRIL 18, 2024
Dr. Mary Talley Bowden recently sued the FDA for stepping beyond their charter, defaming Ivermectin prescribers, and, thereby, interfering with the doctor-patient relationship. Last month, Dr. Bowden resolved her suit, receiving a substantial undisclosed settlement from the government agency.
Dr. Pierre Kory has been an early and staunch defender of the use of Ivermectin to treat COVID-19 in humans. Kory believes the FDA settled this case with Bowden because they had likely hired the PR firm Weber Shandwick to create the now infamous “horse dewormer” campaign (detailed below) to smear Ivermectin and its proponents. If true, once Bowden’s lawsuit went into the phase of discovery then this information would have been revealed, but we will never know since the case is now settled. Weber Shandwick lists the CDC, Pfizer, and Moderna as their clients.
Honest Media covered Ivermectin and the “horse dewormer” controversy in a letter sent to the Associated Press documenting the lies the AP published about the drug. We have also recently received a trove of emails between Dr. Bowden and the Arizona Mirror, an outlet that smeared Dr. Bowden and her colleague, Dr. Peter McCullough. After reviewing them, we can say that these documents illustrate the media’s contempt for medical dissidents.
But why this fear of letting dissenting doctors speak? There has been virtually no coverage of Dr. Bowden’s case. Where there is documentation, like with Jen Christensen’s reporting for CNN, nobody gives voice to the victor and victim, Dr. Bowden. Why?
Dr. Bowden, a Stanford-trained ear, nose, and throat doctor from Houston, has treated more than 6,000 patients suffering from COVID. She is a strong and intelligent woman of science speaking truth to power. Here, in Dr. Bowden, is that “gutsy woman” who Americans were told to admire by leaders like Hillary Clinton. But there’s an implicit caveat in the cult of Clinton’s “gutsy woman:” Such women are to be ignored (and even pilloried and censored) if they challenge the orthodoxies of the Democratic Party or the DNC-aligned Big Pharma industry.
For prescribing Ivermectin and dissenting against the dominant COVID narratives, Dr. Bowden was forced to resign from Houston Methodist Hospital. And she wasn’t the only doctor to face such consequences. Dr. Robert Apter and Dr. Paul Marik, two other Ivermectin physician-advocates, joined Dr. Bowden in her suit against the FDA. Marik, for his part, was forced to resign from Eastern Virginia Medical School as well as Sentara Norfolk General.
Last month, Dr. Bowden traveled to the Supreme Court to stand in solidarity with activists as SCOTUS listened to Murthy v. Missouri. The Murthy case concerns the suppression of medical dissidents, specifically, and online censorship, more broadly. Dr. Bowden addressed the crowd of protesters about her four-year battle with the captured government agency:
How many COVID patients did they examine? How many histories did they take? How many prescriptions did they write? Zero. None of them have cared for a single COVID patient, but because they had the full support of Big Pharma, the government, and, most importantly, the media, they became the scientific authority on a novel disease they had zero first-hand experience in treating.
Bowden has a point. The FDA’s campaign against doctors such as herself gained purchase with the public, in part, because the agency’s claims were amplified by a mainstream media that is shaped and funded – captured – by Big Pharma. Due to the massive influx of advertising dollars and the perfect storm of misinformation and disinformation summoned by Russiagate, the 2020 election, and the COVID-19 pandemic, the American public’s trust in the mainstream media has reached record lows. Bowden’s case reveals another example of why the public is justified in its skepticism.
Let the Doctors Speak
I recently spoke with Dr. Bowden about her fight with the government.
“This was a war on Ivermectin,” she said. “But it was also a war on the doctor-patient relationship.”
I asked her what precipitated the suit against the FDA. Dr. Bowden told me that never before in her career had she witnessed interference with the doctor-patient relationship from the FDA or her local pharmacies. When I asked about prescribing a drug that wasn’t FDA-approved, she told me that she’d often prescribed off-label in the past, with no problems, and that she approached Ivermectin, initially, with hesitancy and skepticism. She said she preferred prescribing monoclonal antibodies at the beginning of the pandemic, but sought new options when access to these treatments became restricted.
“I was nervous to start using it,” she said. “Before I started, I looked at the FDA’s website and the toxicity data. Once I was assured that it worked (maybe not as quickly as monoclonal antibodies), I started offering it to patients.”
Not only did Dr. Bowden prescribe Ivermectin to her patients and witness positive results, but she used it herself. She’s had COVID three times. And in every instance of Ivermectin treatment, both with herself and her patients, she observed either efficacy or minimal side effects.
“I haven’t lost one patient due to Ivermectin,” she said.
In 2015, the Nobel Committee for Physiology honored the discovery of Ivermectin with a Nobel Prize. The NIH lauded this “multifaceted drug,” which was largely unknown in American public discourse prior to the outbreak of the COVID-19 pandemic.
Then, suddenly everyone and their grandmother was an expert on the dangers of Ivermectin. Seemingly overnight, the American people absorbed a viral propaganda campaign from the very government agency (the FDA) that they supported with tax dollars. And if you were a doctor or patient seeking this low-cost, award-winning therapeutic treatment, you were suddenly in the crosshairs of the “war on Ivermectin.” This policing of the poor and the independent all started, according to Dr. Bowden, “with the horse tweet.”
On August 8, 2021, the FDA weaponized its social media account to stigmatize physicians like Dr. Bowden and skeptical and underprivileged patients seeking affordable alternative care. The agency issued a tweet with two images: a veterinarian outdoors caring for a horse, coupled with a physician in an office caring for a masked human. The text for the tweet reads: “You are not a horse. You are not a cow. Seriously, y’all. Stop it.” This tweet, with its careful use of the colloquial and the second person, supplemented with a juvenile binary logic, became the most popular tweet in FDA history.
Hate wins clicks. Fear creates fog. Shortly after the tweet’s publication and viral propagation, Dr. Bowden’s life came undone.
“I never had a pharmacy deny a prescription before,” she said.
Dr. Bowden’s struggle with the pharmacy was just the tip of the iceberg, revealing the stranglehold Big Pharma now has on health care in America. Dr. Bowden suffered (and still suffers) from vicious attacks online, as well as alienation from her peers. She was forced to resign from her workplace, Houston Methodist Hospital. She explained to me that the “war on Ivermectin” was more vitriolic than anything she’d ever seen before in the discourse on public health. And whereas most doctors bent the knee, stayed silent, and complied with government mandates, Dr. Bowden (and others) fought back. Her case represents what one might call a scientific profile in courage.
What does fighting back look like? Well, for starters, perhaps it begins with telling the truth in public and revealing the whole story of Dr. Bowden’s struggle, along with that of fellow medical dissidents like Dr. Kory, Dr. Robert Malone, Dr. Jay Bhattacharya (co-author of the Great Barrington Declaration), and Dr. Peter McCullough.
In Dr. Bowden’s and Dr. McCullough’s recent email exchanges with the Arizona Mirror, one can see, firsthand, a publication that ignores the opportunity to correct factual errors. The Mirror instead willfully litters its reporting on Dr. Bowden and Dr. McCullough with misinformation, ad hominem attacks, bizarre references to Qanon, constant allusions to shadowy conspiracy theories, and the slanderous insinuation that Dr. McCullough is antisemitic.
The Association Fallacy
One of the most recurrent disinformation patterns we have witnessed in studying the defamation of populist voices, broadly, and Dr. Bowden’s case, specifically, is what scholars of rhetoric call the association fallacy. In short, the association fallacy describes claims where even oblique social connection to a stigmatized individual or organization (like QAnon) is used to poison the claims of the targeted speaker. Simply associating the terrifying name of the poisonous organization with the speaker scares the reader and creates an irrational – fallacious – connection.
What’s troubling, in the case of the Arizona Mirror reporting, is that Dr. Bowden and Dr. McCullough have no ties to QAnon. Furthermore, Dr. Bowden and Dr. McCullough both reached out to Jim Small, the paper’s editor, and politely asked that these fallacies be removed from the Mirror’s articles.
For example, Dr. Bowden and Dr. McCullough called attention to the Mirror’s repeated use of the ad hominem “anti-vaxxer” to label Dr. McCullough and associate the doctor with the world of “anti-vaxxers.” In their email exchange, Dr. McCullough confides in Small that he has “accepted dozens of vaccines during the course of my life.”
But the Mirror refused to mirror the truth and remove the slur. The Mirror refused to interview these doctors, refused to correct their reporter’s mistakes when alerted by the victims, and, furthermore, sought to defame the doctors through ad hominem attacks and the association fallacy.
To witness how the association fallacy works, consider the following sentence about Dr. Bowden’s colleague, Dr. McCullough, from the Arizona Mirror’s Jerod Macdonald-Evoy: “McCullough has become a darling to those in both Qanon and the broader conspiracy world, appearing regularly on shows like the one hosted by antisemite Stew Peters, who said the COVID vaccine is a bioweapon.”
In one sentence, the reporter has accused the doctor (without directly accusing him) of antisemitism and conspiracy theory simply by virtue of association with other human beings, mostly unnamed, who populate “the broader conspiracy world.”
What is happening to people like Dr. McCullough and Dr. Bowden rarely happens to those in power. It happens to those who challenge power.
The Arizona Mirror and CNN should be ashamed. They punished informed dissent. They refused to contextualize Dr. Bowden’s struggle as part of a subculture of dignified scientists and physicians. They erased and defamed Dr. Bowden and her colleagues. They published fear porn and called it journalism. They left out this gutsy woman’s voice. Honest Media has chosen a different path. We let the doctor speak.
Fauci’s Inquisition Against Safe and Effective Anti-COVID-19 Drugs

By Richard Gale and Dr. Gary Null | Global Research | April 6, 2024
A question needs to be asked. Were the novel experimental drug treatments for SARS-CoV-2 viral infections that Anthony Fauci, the CDC and FDA advocated for and funded responsible for worsening the contagion and countless deaths?
However, at that time there were plenty of studies confirming there were pre-existing safe, inexpensive medications known to have highly effective antiviral properties to treat Covid-19 patients. Among these were ivermectin and hydroxychloroquine (HCQ).
There were also specific nutrients such as vitamin D and zinc, known to strengthen the immune system against viral infection and yet there was no recommendation from the government about the benefits of proper nutrition. So why did Fauci along with other federal health officials choose to intentionally ignore the scientific evidence and rather condemn these repurposed drugs? In Fauci’s case, over a year and half into the pandemic, he continued to lie outright on CNN that “there is no clinical evidence whatsoever that [ivermectin] works.”[1] And could millions have been saved if these generic medications were prescribed rather than the feds doing nothing but recommending social isolation and quarantines as the world awaited an experimental Covid-19 vaccine to enter the market?
To date, between ivermectin and HCQ alone, there have been 670 published studies, analyses and papers involving over 9,800 scientists and over 682,000 patients supporting the use of these drugs over and beyond those the FDA has approved under Emergency Use Authorization (EUA) statutes. Despite this, four years later, the FDA continues to fiercely deny ivermectin’s and HCQ’s efficacy and safety under proper administration. Why this blatant cover-up?
Every CDC effort to approve a novel drug treatment for SARS-CoV-2 infections has been a dismal failure. Aside from monoclonal antibody therapy, only three anti-Covid-19 drugs have been approved under an EUA in the United States. None met their promised expectations from either the manufacturer or our federal health agencies. With their poor efficacy rates, safety profiles and a black box warning slapped upon Pfizer’s anti-Covid-19 drug Paxlovid, the CDC is scrambling to find new viable alternatives in the pharmaceutical pipeline. Bloomberg amplifies the fake Covid-19 treatment crisis by lamenting that repurposed drugs such as ivermectin are gaining global popularity as “the world needs effective Covid drugs.”[2]
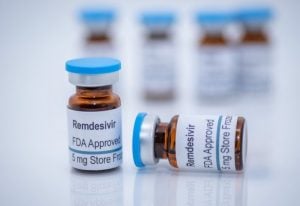
Shortly after the pandemic was formally announced, the FDA recommended the cheap over the counter anti-malarial drug hydroxychloroquine but then quickly reversed its decision after Fauci publicly announced the future arrival of Gilead Sciences’ novel intravenous drug Remdesivir. The FDA’s and European Union’s approvals of Remdesivir baffled many scientists, according to the journal Science, who questioned its therapeutic value and kept a close watch on the drug’s clinical reports about a “disproportionally high number of reports of liver and kidney problems.”[3] Even an earlier Chinese study published in The Lancet found that remdesivir had no impact on the coronavirus. The Science article notes that the “FDA never consulted a group of outside experts that it has at the ready to weigh in on complicated antiviral drug issues.”[4] Six months before remdesivir received EUA approval, Anthony Fauci had already hailed the drug as a major breakthrough that would establish a new “standard of care” in Covid-19 treatment.[5]
Today, remdesivir is being increasingly recognized as a debacle in antiviral therapeutic care. Even the WHO released a “conditional recommendation against the use of remdesivir in hospitalized patients, regardless of disease severity, as there is currently no evidence that remdesivir improves survival and other outcomes in these patients.” An Italian study observed a 416 percent increase in hepatocellular injuries among hospitalized Covid-19 patients treated with Remdesivir.[6] And a smaller Taiwanese study of hospitalized unvaccinated patients reported a 185 percent higher mortality during late remdesivir treatment.[7]
Earlier this year, Pfizer’s novel oral Covid-19 medication Paxlovid was given an FDA black box warning for clinically significant adverse reactions that can potentially be fatal. Because the company does not permit independent random-controlled trials to investigate its drug, other than retrospective studies, we only have Pfizer’s own data to rely upon. Nevertheless, The Lancet published a study by a team of Chinese scientists at Shanghai Jiao Tong School of Medicine that managed to look at Paxlovid’s use among critically ill patients hospitalized with Covid-19. The study reported a 27 percent higher risk of the infection progressing, a 67 percent increased risk in requiring ventilation, and 10 percent longer stays in ICU facilities.[8]
Paxlovid is a combination of a novel SARS-CoV-2 protease inhibitor and the HIV protease inhibitor ritonavir. The FDA approved Paxlovid under a EUA with the claim it was safe. However, on the government’s HIV.gov website for ritonavir it is clearly stated that the drug “can cause serious life-threatening side effects. These include inflammation of the pancreas (pancreatitis), heart rhythm problems, severe skin rash and allergic reactions, liver problems and drug interactions.”[9] Perhaps due to the drug’s serious side effects, it is no longer used solely against HIV, but rather is given in smaller doses as a booster for AZT-related drugs. Being highly toxic, ritonavir is also not recommended for pregnant women and has been shown to interfere with hormone-based birth control efficacy.
Paxlovid only received FDA EUA approval in May 2023. At that time, the agency claimed there was no evidence that patients who were treated with the drug rebounded and came down with Covid. However, shortly thereafter this was determined to be untrue.[10] A Harvard analysis found that 21 percent of Paxlovid recipients will remain contagious and likely succumb to a viral rebound compared to only 1.8 percent who did not take the drug.
Merck’s anti-Covid-19 drug molnupiravir (Lagevrio) also has an FDA black box warning for potential fetal harm when administered to pregnant women. Why the drug was ever approved under an EUA seems to be an enigma. The drug’s antiviral activity is based upon a metabolite known as NHC, which for many years has been known to create havoc in an enzyme crucial for viral replication by inserting errors into the virus’ genetic code. The theory is: produce enough errors and the virus kills itself off. However, molnupiravir can cause hundreds of mutations thereby “supercharging” the manufacturing of new Covid-19 viral strains. Moreover, according to a Forbes article, the drug’s mutagenic powers may also interfere with our own body’s enzymes and DNA.[11] Another Forbes article points out that Merck’s clinical trial only enrolled around 1,500 participants, which is far too “small to pick up on rare mutagenic events.”[12]
Molnupiravir has a poor efficacy rate across the board including viral clearance, recovery, and hospitalizations/death (68 percent).[13] One trial, funded by Merck, concluded the drug had no clinical benefit.[14] More worrisome, the drug also has life-threatening adverse effects including mutagenic risks to human DNA and mitochondria, carcinogenic activity and embryonic death.[15]

Each of these drugs have been outrageous cash cows for their manufacturers. Remdesivir is priced at $3,120 per treatment and earned Gilead $5.6 billion in sales for 2021.
Pfizer’s Paxlovid is priced at $1,390 per treatment. Last year, the company’s revenues for its Covid products—Paxlovid and the Comirnaty vaccine—came in at $12.5 billion, and, according to Fierce Pharma, Pfizer wrote off an additional $4.7 billion on its overstocked Paxlovid inventory.[16] Merck’s molnupiravir’s sales for 2022 cashed in almost $5.7 billion. Despite their profits, none of these drugs have been shown convincingly to have measurably lessened the pandemic nor the spread of SARS-CoV-2.
Despite all the attention and medical hype about novel experimental antiviral drugs to treat Covid-19, Anthony Fauci and other federal officials had full knowledge that other FDA-approved drugs existed that could have been quickly repurposed at minimal expense to effectively treat Covid-19 infections. Repurposing existing drugs to treat illness is a common occurrence. The antiparasitic and antiviral drug Ivermectin best stands out. Its effectiveness was observed to be so remarkable and multifaceted that researchers started to investigate its potential.
The mainstream media, including many liberal news sources who pride themselves on their independence, continue to channel the voices of Anthony Fauci, the CDC and FDA to demonize ivermectin and other generic drugs for treating Covid-19 and to reduce hospitalization and deaths. This propaganda campaign, however, has completely ignored the large body of medical literature that shows ivermectin’s statistically significant efficacy against symptomatic and asymptomatic SARS-2 infections.
Originally developed for veterinarian use, in 1987, the FDA approved ivermectin for treating two parasitic diseases, river blindness and stronglyoidiasis, in humans. Since then an enormous body of medical research has grown showing ivermectin’s effectiveness for treating other diseases. Its broad range of antiviral properties has shown efficacy against many RNA viruses such avian influenza, zika, dengue, HIV, West Nile, yellow fever, chikungunya and earlier severe respiratory coronaviruses. It has also been shown to be effective against DNA viruses such as herpes, polyomavirus, and circovirus-2.[17]
Unsurprisingly, ivermectin’s inventors Drs. William Campbell and Satoshi Omura were awarded the 2015 Nobel Prize in Physiology and Medicine.
It has been prescribed to hundreds of millions of people worldwide. Given its decades’ long record of in vitro efficacy, it should have been self-evident for Fauci’s NIAID, the CDC and the WHO to rapidly conduct in vivo trials to usher ivermectin as a first line of defense for early stage Covid-19 infections and for use as a safe prophylaxis.
For example, if funding were devoted for the rapid development of a micro-based pulmonary delivery system, mortality rates would have been miniscule and the pandemic would have been lessened greatly.[18] Repurposing ivermectin could have been achieved very quickly at a minor expense.[19] However, despite all the medical evidence confirming ivermectin’s strong antiviral properties and its impeccable safety record when administered properly, we instead witnessed a sophisticated government-orchestrated campaign to declare war against ivermectin and another antiviral drug, hydroxychloroquine (HCQ), in favor of far more expensive and EUA approved experimental drugs. Unlike the US, other nations were eager to find older drugs to repurpose against Covid-19 and protect their populations. A Johns Hopkins University analysis offered the theory that a reason why many African countries had very few to near zero Covid-19 fatalities was because of widespread deployment of ivermectin. In February 2020, the National Health Commission of China, for example, was the first to include hydroxychloroquine in its guidelines for treating mild, moderate and severe SARS-2 cases. Eight Latin American nations distribute home Covid-19 treatment kits that include ivermectin.[20] Why did the US and most European countries swayed by the US and the WHO fail to follow suit?
Early in the pandemic, physicians in other nations where treatment was less restricted, such as Spain and Italy, shared data with American physicians about effective treatments against the SARS-2 virus. In addition, there was a large corpus of medical research indicating that older antiviral drugs could be repurposed. Doctors who started to prescribe drugs such as ivermectin and HCQ, along with Vitamin D and zinc supplementation, observed remarkable results. Unlike the dismal recovery and high mortality rates reported in hospitals and large clinics that relied upon strict isolation, quarantine, and ventilator interventions, this small fringe group of physicians reported very few deaths among their large patient loads. Even reported deaths were more often than not compounded by patients’ comorbidities, poor medical facilities and other anomalies.
Very early into the pandemic, medical papers indicated ivermectin was a highly effective drug to treat SARS-2 infections.
In April 2020, less than a month after the WHO declared Covid-19 as a global pandemic, Australian researchers at the Peter Doherty Institute of Infection and Immunity published a paper demonstrating that a single ivermectin dose can control SARS-CoV-2 viral replication within 24-48 hours.[21] Monash University’s Biomedicine Discovery Institute in Australia had also published an early study that ivermectin destroyed SARS-2 infected cell cultures by 99.8 percent within 48 hours. But no American federal health official paid any attention.
As of March 2024, a database for all studies and trials investigating ivermectin against Covid-19 infections records a total of 248 studies, 195 peer-reviewed, and 102 involving controlled groups reporting an average 61 percent improvement for early infections, a 39 percent success rate in treating late infections, and an 85 percent average success rate for use as a preventative prophylaxis.[22] Moreover, prescribing ivermectin reduced mortality by 49 percent, compared to remdesivir’s 4 percent, Pfizer’s Paxlovid’s 31 percent, and molnupiravir’s 22 percent. Even hydroxychloroquine well outperforms these drugs mortality risk for early treatment at 66 percent.
A noteworthy study conducted in Brazil and published in the Cureus Journal of Medical Science prescribed ivermectin in a citywide prophylaxis program in a town of 223,000 residents. 133,000 took ivermectin. The results for a population of this size are indisputable in concluding that ivermectin is a safe first line of defense to confront the pandemic. Covid mortality was reduced 90 percent. There was also a 67 percent lower risk of hospitalization and a 44 percent decrease in Covid cases. Garcia-Aquilar et al reports a Mexican in vitro analysis showing a definitive interaction between ivermectin and the SAR-CoV-2 spike protein, which would account for its high efficacy in Covid-19 cases.[23]
The All India Institute for Medical Science (AIIMS) and the Indian Council of Medical Research (ICMR), two of India’s most prestigious institutions, acted against the WHO and launched an ivermectin treatment campaign in several states. In Uttar Pradesh there was a 95 percent decrease in morality (a decline from 37,944 to 2,014). The Indian capital of New Delhi witnessed a 97 percent reduction. During the same time period, the state of Tamil Nadu, which followed the WHO’s ban on ivermectin, had a 173 percent increase in deaths (from 10,986 to 30,016 deaths).
There have been many concerted efforts to discredit ivermectin and other repurposed drugs’ effectiveness. Most notable is the large TOGETHER Trial Brazil study published in the New England Journal of Medicine (NEJM) that concluded both ivermectin and another repurposed drug fluvoxamine showed no beneficial signs for treating Covid-19 patients. The study was widely reported in the mainstream media. However, a Cato Institute analysis discovered the study in fact showed its benefits and the results were in agreement with 87 percent of other clinical trials investigating ivermectin. The Cato analysis identifies many odd anomalies in how the trial was conducted including an unspecified placebo—although it is suspected it was Vitamin C, which has itself been shown to be mildly effective against the SARS-CoV-2 virus, and protocol changes as the study was underway including inclusion/exclusion criteria. By his own admission the TOGETHER Trial’s principal investigator Dr. Ed Mills at McMaster University in Ontario “designs clinical trials, predominantly for the Bill and Melinda Gates Foundation.”[24] In a McMaster University press release, the Gates foundation is listed as a funder for the study to debunk ivermectin and fluvoxamine.[25] Oddly, Gates is nowhere listed among the several funders in the NEJM study’s disclosure. In addition, TOGETHER Trials is owned by the Canadian for profit startup Purpose Life Sciences, founded by Mills; legal documents showed Mills’ PLS is largely funded and controlled by Sam Bankman Fried’s FTX who invested $53 million into the project. Administrators of FTX’s bankruptcy are suing PLS for fraud.[26]
In short, the ivermectin/fluvoxamine TOGETHER Trial was a complete medical sham and intentionally designed for one single purpose: to fuel media disinformation in order to undermine ivermectin’s superior efficacy and safety profile to Big Pharma’s more profitable designer drugs.
In 2004, the US Congress passed an amendment to the Federal Food, Drug and Cosmetic Act known as Emergency Use Authorization (EUA). This piece of legislature legalized an anti-regulatory pathway to allow experimental medical interventions to be expedited and bypass standard FDA safety evaluations in the event of bioterrorist threats and national health emergencies such as pandemics. At the time, passage of the EUA amendment made sense because it was partially in response to the 2001 anthrax attacks and the US’s entry into an age of international terrorism. However, the amendment raises some serious considerations. Before the Covid-19 pandemic, EUAs had only been authorized on four occasions: the 2005 avian H5N1 and 2009 H1N1 swine flu threats, the 2014 Ebola and the 2016 Zikra viruses. Each of these pathogen scares proved to be false alarms that posed no threat of pandemic proportions to Americans. The fifth time EUAs were invoked was in 2020 during the Covid-19 pandemic, which at the time seemed far more plausible.
Before the government can authorize an EUA to deploy an experimental diagnostic product, drug or vaccine, certain requirements must be fulfilled. First, the Secretary of the Department of Health and Human Services (HHS) must have sufficient proof that the nation is being confronted with a serious life-threatening health emergency. Second, the drug(s) and/or vaccine(s) under consideration must have sufficient scientific evidence to suggest they will likely be effective against the medical threat. The evidence must at least include preclinical and observational data showing the product targets the organism, disease or condition. Third, although the drug or vaccine does not undergo a rigorous evaluation, it must at least show that its potential and known benefits outweigh its potential and known risks. In addition, the product must be manufactured in complete accordance with standard quality control and safety assurances.
When we look back at the government’s many debacles during the Covid-19 pandemic, other EUA requirements warrant the spotlight. On the one hand, an EUA cannot be authorized for any product or intervention if there is an FDA alternative approved product already available, unless the experimental product is clearly proven to have a significant advantage. Moreover, and perhaps more important, EUAs demand informed consent. Every individual who receives the drug or vaccine must be thoroughly informed about its experimental status and its potential risks and benefits. Recipients must also be properly informed about the alternatives to the experimental product and nobody should be forced to take it.
Finally, an EUA requires robust safety monitoring and reporting of adverse events, injuries and deaths potentially due to the drug or vaccine. This is the responsibility not only of the private pharmaceutical manufacturers but also the FDA, physicians, hospitals, clinics and other healthcare professionals.
Obviously important cautions must be considered after approving a medical intervention under the EUA requirements. Foremost are the inherent health risks of any rapid response of experimental medical interventions, especially novel drugs and vaccines. As we observed during the FDA approval process and roll out of Pfizer’s and Moderna’s mRNA Covid-19 jabs, no long-term human trials were conducted to even estimate a reliable baseline of their relative efficacy and safety. The American public has blindly placed its trust in our federal health authorities decision-making. It is expected that under a national health emergency, the authorities would be completely transparent and act only by the highest ethical standards. However our institutions betrayed public trust and either ignored or transgressed cautions underlying EUA approved medical interventions in every conceivable way. Moreover, conflicts of interests have been discovered to have plagued the entire EUA review process.
Although the EUA amendment provides some protections to authorized drug and vaccine manufacturers, it was the Public Readiness and Emergency Preparedness Act (PREP) in 2005 that expanded liability protections. In addition to protecting private corporations, PREP also shields company executives and employees from claims of personal injury or death resulting from the administration of authorized countermeasures. The only exceptions for liability are if the company or its executive offices are proven to have engaged in intentional and/or criminal misconduct with conscious disregard for the rights and safety of those taking their drugs and vaccines.
During the pandemic, the FDA issued widespread EUAs with liability immunity for the PCR diagnostic kits for SARS-2, the mRNA vaccines and the anti-Covid-19 drugs. Curiously, the Secretary of the Department of Health and Human Services invoked the PREP Act on February 4, 2020 giving liability protections; this was over a month before the pandemic was officially announced, which raises serious questions about prior-planning before the viral outbreak in Wuhan, China.
From the pandemic’s outset, Fauci embarked on the media circuit to promise Americans that federal health agencies were doing everything within their means to get a vaccine on the market because there was no available drug to clear the SARS-2 virus. As we have seen with respect to ivermectin alone, this was patently false. Rather the government placed an overriding emphasis on vaccination with a near total disregard for implementing very simple preventative measures to inhibit viral progression. Once mass vaccinations were underway, we were promised that the SARS-2 virus would be defeated and life would return to normal. In retrospect, we can look back and state with a degree of certainty that American health authorities and these products’ corporate manufacturers may have violated almost every EUA requirement. Everything that went wrong with the PCR kits, the experimental mRNA vaccines and novel drugs could have been avoided if the government had diligently repurposed effective and safe measures as pandemic countermeasures. Very likely, hundreds of thousands of lives, perhaps millions, would have been saved.
Similarly the FDA issued a warning statement against the use of ivermectin. Even ivermectin’s manufacturer Merck discredited its own product. Shortly after ridiculing its drug, the Alliance for Natural Health reported, “Merck announced positive results from a clinical trial on a new drug called molnupiravir in eliminating the virus in infected patients.”[27]
And still the FDA considers these novel patented drugs to be superior to ivermectin. Favoring a vaccine regime and government-controlled surveillance measures to track every American’s movements, American health officials blatantly neglected their own pandemic policies’ severe health consequences. Ineffective lockdowns, masks, social isolation, unsound critical care interventions such as relying upon ventilators, and the sole EUA approvals of the costly and insufficiently effective drugs brought about nightmares for tens of millions of adults and children. This was all undertaken under Fauci’s watch and the heads of the US health agencies in direct violation of the EUA requirements to only authorize drugs and medical interventions when no other safe and effective alternative is available. Alternatives were available.
The 4-year history of the pandemic highlights a sharp distinction between dependable medical research and pseudoscientific fraud. The CDC adopted a common Soviet era practice to redefine the very definition of a vaccine and the parameters of vaccine efficacy in order to fit economic and ideological agendas. This explains Washington’s aggressive public relations endeavors to silence medical opponents. According to cardiologist Dr. Michael Goodkin’s private investigations, several of the most cited studies discrediting ivermectin’s antiviral benefits were intentionally manipulated in order to produce “fake” results.[28] These studies were then widely distributed to the AMA, American College of Physicians and across mainstream media to author “hit pieces” to demonize ivermectin and other repurposed drugs. The government’s belligerent and reactive diatribes, brazenly or casually advocating for censorship, were direct violations of scientific and medical integrity and contributed nothing towards developing constructive policies for handling a pandemic with a minimal cost to life. The consequence has been a less informed and grossly naïve public, which was gaslighted into believing lies.
The FDA’s EUAs for the Covid-19 vaccines and novel experimental drugs were in fact an attack on the amendments and PREP directives. Neither the vaccines nor drugs warranted emergency authorization because effective and safe alternatives were readily available. No doubt a Congressional investigation would uncover criminal misconduct and conscious fraud. Moreover, these violations of the PREP Act may have the potential to lead directly into medical crimes against humanity as outlined in the Nuremberg Code.
Although the Nuremberg Code has not been officially adopted in its entirety as law by any nation or major medical association, other international treaties, such as the Universal Declaration of Human Rights, the World Medical Association Declaration of Helsinki (which is not legally binding), the International Covenant on Civil and Political Rights (ICCPR) and the International Ethical Guidelines for Biomedical Research on Human Subjects incorporate some of Nuremberg’s main principles that aim to protect people from unethical and forced medical research. Although the US signed the ICCPR as an intentional party, the US Senate never ratified it. The ICCPR’s Article 7 clearly states, “No one shall be subject to torture or cruel, inhuman or degrading treatment or punishment,” which can legally be interpreted to include forced medical experimentation implied as cruel, inhuman treatment. Other ICCPR articles, 6 and 17, are also applicable to medical experimentation to ensure ethical conduct, obtaining proper informed consent and the right to life and privacy. For a moment, consider the numerous senior citizens in nursing homes and hospitals who were simply administered experimental Covid-19 vaccines without full knowledge about what they were receiving. And now how many children are being coerced by the pseudoscience of health officials’ lies to be vaccinated without any knowledge of these mRNA products’ risk-benefit ratio?
The US is also a signatory to the Helsinki Declaration, which, although not directly aligned with Nuremberg, shares much in common. The Declaration shares some common features with the EUA amendment and PREP Act. These include voluntary informed consent—which is universally accepted, adequate risk and benefit information about medical interventions, and an emphasis on the principle of medical beneficence (promoting well-being and the Hippocratic rule of doing no harm). It also guarantees protections for vulnerable groups, especially pregnant women and children, which the US government and vaccine makers directly violated by conducting trials on these groups with full knowledge about these vaccines’ adverse events in adults. In addition, weighing the scientific evidence to assess the risk-benefit ratios between prescribing ivermectin and HCQ over the new generation of novel experimental drugs conclusively favors the former. This alone directly violates the ethical medical principles noted above.
However, the failure to repurpose life-saving drugs is less criminal than the questionable unethical motivations to usher a new generation of genetically engineered vaccines that have never before been adequately researched in human trials for long term safety. This mass experimentation, which continues to threaten the health and well-being of millions of people, is global and can legally be interpreted as a genocidal attack on humanity.
If the emerging data for increasing injuries and deaths due to the Covid-19 vaccines is reliable—and we believe it is—the handling of the pandemic can be regarded as the largest medical crime in human history. In time, and with shifting political allegiances and public demands to hold our leaders in government and private industry accountable, the architects of this medical war against civilization will be brought to justice.
*
Richard Gale is the Executive Producer of the Progressive Radio Network and a former Senior Research Analyst in the biotechnology and genomic industries.
Dr. Gary Null is host of the nation’s longest running public radio program on alternative and nutritional health and a multi-award-winning documentary film director, including his recent Last Call to Tomorrow.
Notes
[1] https://www.cnn.com/videos/health/2021/08/29/dr-anthony-fauci-ivermectin-covid-19-sotu-vpx.cnn
[4] https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(20)31022-9/fulltext
[6] https://www.dldjournalonline.com/article/S1590-8658(21)00923-3/fulltext
[8] https://www.thelancet.com/action/showPdf?pii=S2666-6065%25252823%25252900012-3
[9] https://clinicalinfo.hiv.gov/en/drugs/ritonavir/patient
[10] https://www.yalemedicine.org/news/13-things-to-know-paxlovid-covid-19
[13] https://www.medrxiv.org/content/10.1101/2023.01.20.23284849v1.full.pdf
[14] https://evidence.nejm.org/doi/pdf/10.1056/EVIDoa2100044
[15] https://c19early.org/waters.html
[17] https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7290143/
[18] https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7539925/
[19] https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7564151/
[21] https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7129059/
[22] https://c19ivermectin.com
[23] https://www.mdpi.com/1422-0067/24/22/16392
[25] https://www.eurekalert.org/news-releases/855535
[26] https://c19ivm.org/tallaksen.html
[27] https://anh-usa.org/fda-ensures-pharma-profits-on-covid/
[28] https://www.trialsitenews.com/a/are-major-ivermectin-studies-designed-for-failure
FDA is trying to legalize its illegal approvals of COVID vaccines without human trials
Now one animal trial will do the trick to “prove” efficacy?

BY MERYL NASS | MARCH 26, 2024
You can’t prove efficacy with an animal trial. You can’t prove safety with an animal trial. The anthrax vaccine studies PROVED that even when a vaccine seemed to work in animals, it was unclear what the right dose was and equally unclear how antibody levels related to protection, because they didn’t. Maybe because no one knew which antibody to measure?
Surrogate markers for approval of vaccines (or anything else) became a total joke when FDA began ignoring the requirement that “correlates of protection” were a essential before you could use antibodies, T cells, animals or anything besides humans to assess a vaccine. This means that you have to prove the surrogate marker or animal model actually duplicates the human response, since usually they don’t.
FDA officials ditched this absolutely necessary standard (imho illegally) and now want to a) cover their tracks, and b) foist more untested and possibly deadly injections on us going forward.
Who needs them? They are simply a Potemkin Village of a regulatory agency at this point. Thankfully ICAN is watching.
With public faith in FDA continuing to decline, the agency published draft guidance stating that a single animal study could be sufficient to demonstrate a vaccine’s effectiveness. ICAN’s attorneys submitted a comment in opposition to this draft guidance which comes shortly after FDA approved a COVID-19 booster based on trials performed solely in mice.
Back in 2007, an FDA advisory committee met to re-review the safety, effectiveness, and dosing of phenylephrine (Sudafed PE), a drug which has been on the market for nearly 50 years. At the 2007 meeting, the committee considered its efficacy data “borderline” but nonetheless voted 11-1 in favor of its efficacy, although it did vote in favor of additional studies being done.
On September 12, 2023, the same FDA committee, possibly attempting to bolster its image as a tough regulatory watchdog, voted unanimously to label phenylephrine “ineffective” as a nasal decongestant. The committee noted “significant methodological and statistical issues with the design and conduct of the original studies submitted to and evaluated by the Panel… All but one evaluated extremely small sample sizes, none adequately controlled for bias … and none performed appropriate sample size calculations,” and observed, “After a thorough review of all the available evidence, it is also possible that there may have been bias and/or data integrity issues at [at] least one study center … where five of the seven positive oral PE studies were conducted.”
If FDA follows the panel’s advice, the drug will be pulled from the market, despite the long-standing indications of its inefficacy, most of which were known in 2007. It’s too bad the committee didn’t perform this “thorough review” at any point prior to the last 50 years, before Americans wasted billions of dollars on an ineffective treatment (nearly $2 billion in 2022 alone) that apparently only exposed them to potential harm without benefit.
Ironically, on September 11, 2023, just one day before the committee met to clamp down on this “ineffective drug” with shoddy clinical trials, FDA issued draft guidance stating that, under certain circumstances, a manufacturer can show evidence of effectiveness for a biological product with a single clinical investigation conducted in animals, giving the example of “[w]hen the product is a preventive vaccine, and there is a well-established model of infection for a relevant infectious disease, and use of the vaccine in the animal model demonstrates prevention of disease.” Notably, this occurred shortly after FDA authorized the newest Pfizer COVID-19 vaccines, which are as Dr. Paul Offit notes, “new product[s],” even though Pfizer tested this brand-new shot only in mice — not humans.
Thus, on December 18, 2023, ICAN, through its attorneys, filed a formal comment in opposition to the draft guidance. In it, ICAN points out that, despite urging by ICAN in its November 2020 petition, FDA refused to amend the Phase III trials of the COVID-19 vaccines to ensure they met the required standard of “substantial evidence” of effectiveness. As a result, we now have generations of new vaccines being approved based on those original ineffective products.
Now, FDA is doubling down on its malfeasance by authorizing COVID-19 vaccines without human trials while simultaneously creating guidance to excuse its lack of oversight. As ICAN’s comment notes:
[I]t is apparent that FDA is tailoring guidance based on the vaccine manufacturers’ clinical trials instead of requiring that these trials comply with what any reasonable licensing agency should and would require. FDA has seemingly forgotten that its function is to regulate the pharmaceutical industry, not rubber stamp it. The Draft Guidance does not provide oversight, and even worse lends illegitimacy to the FDA when there is clearly insufficient evidence to support authorization or licensure. This is especially troubling for products that will be injected into healthy humans including babies, children, and pregnant women.
Rest assured that ICAN’s legal team will continue to serve as a watchdog for FDA and ensure it is held accountable for improperly wielding its regulatory authority and fulfilling its responsibilities.
To support future legal action like this, click here to donate!
You can read more about ICAN’s work serving as watchdog for our federal health agencies at the links below:
Sen. Ron Johnson Accuses Public Health Agencies of ‘Appalling Lack of Transparency’ on COVID Vaccines
By Brenda Baletti, Ph.D. | The Defender | October 27, 2023
Sen. Ron Johnson (R-Wis.) accused federal public health agencies of displaying an “appalling” lack of transparency with the American public during the pandemic, depriving them of “the benefit of informed consent.”
In a letter sent Oct. 25 to the heads of the U.S. Department of Health and Human Services, the U.S. Food and Drug Administration (FDA), the Centers for Disease Control and Prevention (CDC) and the National Institutes of Health, Johnson said that even now, “As new and alarming information continues to come to light, federal health agencies continue to stonewall and gaslight Congress and the public.”
The lawmaker pointed to an FDA-funded study published this month that identified a potential safety signal linking mRNA COVID-19 vaccines to seizures in children ages 2-5.
Johnson questioned whether the CDC was aware of these findings last month when it recommended everyone 6 months of age and older be vaccinated to protect against COVID-19 this fall and winter.
Johnson said the leaders of these agencies — Xavier Becerra, Dr. Robert Califf, Dr. Mandy Cohen, and Lawrence Tabak, D.D.S., Ph.D. — have failed in their duty to be transparent with Americans regarding what they knew about the safety and efficacy of the vaccines.
As a result, they “have not even come close to ensuring that doctors can provide informed consent on a new gene therapy masquerading as a ‘vaccine’ that was rushed to market without adequate safety or efficacy testing,” he said.
The Wisconsin senator has been a vocal critic of the federal COVID-19 response and an outspoken advocate for people injured by the vaccine. In 2022, he led a roundtable discussion with doctors and scientists to shed light on what was known so far about the vaccines.
Johnson has also accused the CDC of colluding with Twitter to censor his own social media posts about the vaccines.
In his letter, the lawmaker wrote that the agencies’ refusal to respond to the “vast majority” of his questions and information requests “only heightens [his] level of suspicion.”
He listed over a dozen letters he sent requesting information on the COVID-19 vaccines that the agencies “have failed to adequately address.”
These included requests for data about vaccine lots linked to high rates of adverse events, information suppression on social media and the Countermeasures Injury Compensation Program.
The CDC and FDA also failed to fulfill Johnson’s requests for their adverse events surveillance data and their analyses of the Vaccine Adverse Event Reporting System, or VAERS, database.
Children’s Health Defense also is suing the FDA to respond to its Freedom of Information Act requests to make that same data available.
Johnson listed 11 other outstanding requests he made regarding the other aspects of the pandemic.
But these make up only a partial list of over 60 public letters Johnson said he has sent to government agencies concerning various aspects of the pandemic.
“It is well past time for U.S public health agencies to be transparent,” he said.
Johnson requested the agencies respond by Nov. 8 to questions about what they knew about the risks COVID-19 vaccines posed to children, when they knew it and how they plan to address those issues.
Brenda Baletti Ph.D. is a reporter for The Defender. She wrote and taught about capitalism and politics for 10 years in the writing program at Duke University. She holds a Ph.D. in human geography from the University of North Carolina at Chapel Hill and a master’s from the University of Texas at Austin.
This article was originally published by The Defender — Children’s Health Defense’s News & Views Website under Creative Commons license CC BY-NC-ND 4.0. Please consider subscribing to The Defender or donating to Children’s Health Defense.
FDA ties with Gates Foundation
Maryanne Demasi, reports | October 4, 2023
In 2017, the US Food and Drug Administration (FDA) entered into a memorandum of understanding (MOU) with the Bill & Melinda Gates Foundation.
Under the MOU, the two entities agreed to share information to “facilitate the development of innovative products, including medical countermeasures,” such as diagnostics, vaccines, and therapeutics to combat disease transmission during a pandemic.
The FDA has MOUs with many academic and non-profit organisations, but few have as much to gain as Bill Gates, who has invested billions into pandemic countermeasures.
Experts are concerned the Gates Foundation could have undue influence over the FDA’s regulatory decisions of these countermeasures.
David Gortler, an ex-senior adviser to the FDA commissioner between 2019 and 2021, says he is “suspicious” of the MOU.
“If the Gates Foundation establishes an MOU with a regulator on a product they want to develop, it seems like it would be a conflict of interest. What if every other drug company did the exact same thing as the Gates Foundation?” he says.

David Gortler, former senior advisor to FDA commissioner 2019-2021
Gortler, now a fellow at the Ethics and Public Policy Center in Washington DC, explained that normally, meetings between developers and regulators are supposed to be an official part of the public record and subject to Freedom of Information Act requests.
“However, an MOU such as this can circumvent the usual requirements for the transparency of official communications,” says Gortler. “This way their communications can be kept secret.”
David Bell, a former medical officer for the World Health Organisation (WHO) who now works as a public health physician and biotech consultant, agrees that the MOU has potential to corrupt the regulatory process.
“The narrative is that philanthropic foundations can only be good, because they’re making vaccines and saving thousands of lives, so we need to cut the red-tape and help the FDA get stuff done quickly otherwise children will die,” says Bell. “But in reality, it has potential to corrupt the whole system.”

David Bell, physician and biotech consultant
Bell adds, “Speaking generally, close relationships between regulators and developers raise inevitable risks that shortcuts and favours will break down the rigorousness of the product review, putting the public at risk.”
Revolving door
The FDA has been roundly criticised for its “revolving door.” Ten of the past 11 FDA commissioners left the agency and secured roles with pharmaceutical companies they once regulated.
Similarly, the Gates Foundation hired high-ranking members of the FDA, who bring with them intimate knowledge of the regulatory process.
For example, Murray Lumpkin had a 24-year career at the FDA, serving as senior advisor to the FDA commissioner and representative for global issues. Now, he is deputy director of regulatory affairs at the Gates Foundation, and signatory on the MOU.
And Margaret Hamburg, who served as FDA commissioner between 2009 and 2015, is now on the Scientific Advisory Board of the Gates Foundation.

Murray Lumpkin, deputy director regulatory affairs, Gates Foundation; Margaret Hamburg, scientific advisory board, Gates Foundation
Bell has no doubt that these appointments were strategic to “game the system” saying, “If I worked at the Gates Foundation, I would certainly hire somebody like Murray Lumpkin.”
The only way to fix the revolving door problem Bell says, is to have a ‘non-compete clause’ in their contracts.
“It might be that FDA employees cannot work for the people they’ve regulated for at least 10 years. There are places that have those rules – private companies have agreements that you can’t work for a rival,” said Bell.
The FDA dismissed questions about the potential for conflicts of interest, or the lack of transparency over its communications with the Gates Foundation. In a statement, the FDA said:
FDA regulatory decision making is science-based. Former FDA officials do not impact regulatory decisions. FDA only collaborates with the Bill and Melinda Gates Foundation under the MOU as described.
Gates has billions at stake
Gates boasted about receiving a 20-to-1 return on his $10 billion investment into the “financing and delivery” of medicines and vaccines.
“It’s the best investment I’ve ever made,” he wrote in The Wall Street Journal. “Decades ago, these investments weren’t sure bets, but today, they almost always pay off in a big way.”
In Sept 2019, just prior to the pandemic, SEC filings showed the foundation purchased over 1 million shares for $18.10/share. By Nov 2021, the foundation dumped most of the stock for an average of $300/share.
Investigative journalist Jordan Schachtel reported the foundation pocketed approximately $260 million in profit – more than 15 times its original investment – most of it untaxed because it was invested through the foundation.
In his recent book, “How to Prevent the Next Pandemic,” Gates warns that future pandemics are the biggest threat to humankind and that survival depends on global pandemic preparedness strategies, firmly positioning himself at the centre of shaping the agenda.
In October 2019, the Gates Foundation and the World Economic Forum hosted Event 201, which gathered government agencies, social media companies and national security organisations to war game a “fictional” global pandemic.

October 2019, Gates and WEF fund Event 201 to simulate a global pandemic response
The key recommendations from the event were that such a crisis would require the deployment of new vaccines, surveillance and control of information and human behaviours, by orchestrating the co-operation and co-ordination of key industries, national governments, and international institutions.
Several weeks later when the covid-19 pandemic emerged, many aspects of this ‘hypothetical scenario’ became a chilling reality.
The Gates Foundation, which holds shares in a range of drug companies including Merck, Pfizer, and Johnson & Johnson, is now credited with wielding significant influence over the direction of the global response to the pandemic, saying its goal is to “vaccinate the entire world” with a covid-19 vaccine.
Global dominance
The Gates Foundation has poured millions into funding NGOs, media, and international agencies, earning Gates significant political clout.
Financial contributions to the media have garnered Gates favourable news coverage, boasting on the foundation’s website it committed almost $3.5 million to The Guardian in 2020 – 2023.
The UK medicines regulator – the MHRA – disclosed it took approximately $3 million in funding from the Gates Foundation in 2022, which would span across several financial years.
Presidential candidate Robert F Kennedy Jr labelled Gates “the most powerful man in public health” because he managed to steer the WHO’s pandemic strategy to focus primarily on vaccination.
Kennedy said in an interview that the WHO “begs and rolls over” for Gates’ funding, which now makes up over 88% of the total amount of the WHO’s donations by philanthropic foundations.

Robert F Kennedy Jr, Presidential Candidate
“I think [Gates] believes that he is somehow ordained divinely to bring salvation to the world through technology,” said Kenney. “He believes the only path to good health is inside a syringe.”
The Gates Foundation’s CEO Mark Suzman responded to concerns that the foundation has “disproportionate sway in setting national and global agendas, without any formal accountability to voters or international bodies.”
“It’s true that between our dollars, voice, and convening power, we have access and influence that many others do not,” admitted Suzman in his 2023 annual letter .
“But make no mistake – where there’s a solution that can improve livelihoods and save lives, we’ll advocate persistently for it. We won’t stop using our influence, along with our monetary commitments, to find solutions,” he wrote.
FDA rush to approve killer ‘vaccines’ but reject life-saving allergy treatment
By Roger Watson | TCW Defending Freedom | September 25, 2023
The US Food and Drug Administration (FDA), quick to approve an experimental and untested Covid-19 ‘vaccine’, is now stalling over approval of a new device that could save the lives of people who go into anaphylactic shock.
Anaphylactic shock, or anaphylaxis, is an extreme immune system reaction to an allergy which leads to a massive release of chemicals around the body, principally histamine, in an effort to deal with an allergen. This reaction of the body is, in fact, an overreaction and the response leads to difficulties with breathing and sudden and dramatic drop in blood pressure (shock). If untreated it can be fatal.
One of the first lines of treatment for anaphylaxis is an EpiPen which can be carried by those who know they have allergies. It is a device containing the drug noradrenaline (called norepinephrine in the US) that counteracts the shock induced by anaphylaxis. However, an EpiPen requires the injection of noradrenaline intramuscularly. This is invasive and if the person is unable to administer the drug others can be reluctant, either not knowing how to use the device or being afraid to. Also, the drug has to get from the musculature to the bloodstream to be effective. This can take five to ten minutes during which time the person may die.
How much better if noradrenaline could be administered more directly into the bloodstream. Manufacturing a device to do this intravenously, far less expecting anyone other than a medic, nurse or paramedic to have the expertise to do this in an emergency, is a tall order. But drugs can be administered rapidly into the bloodstream nasally and a company, ARS Pharmaceuticals, has developed an injection-free device for administering noradrenaline nasally in the form of an epinephrine nasal spray.
What’s not to like, you may think. Well, the FDA don’t like it and have withheld approval pending further testing. The FDA objections are based on the need for further clinical data in people with anaphylaxis due to the fear that it may fail to work.
It is, surely, heartening that the FDA has the best interests of potential anaphylaxis sufferers at heart. If only they had had the same concern on 2021 when they approved the Pfizer Covid-19 vaccines. It has been exposed in these pages from the start of the Covid-19 vaccine rollout that these were ‘experimental’ medications and had not been tested adequately before approval. Well, they have been tested now and the outcome is clear: they were completely unnecessary; they do not work; and they are extremely dangerous leading to injury and death. They undoubtedly have played a large part in the level of excess deaths observed in recent years.
According to UK government figures, the overall fatality rate from Covid-19 (always a disputed figure due to the difficulty in distinguishing ‘with Covid’ from ‘from Covid’) is 0.1 per cent. The estimated fatality rate from anaphylaxis – made uncertain by the fact that many suspected cases are dead before being found – is between 0.7 and 2 per cent. Recent England hospital Covid-19 admissions are approximately 3,000 per month at the latest available figures. The annual rate of hospitalisation in England for anaphylaxis in 2022-23 was 26,000; at approximately 2,000 monthly, the same order of magnitude as the Covid-19 admissions.
Bearing in mind that it is not easy to distinguish Covid-19 from other respiratory infections but that anaphylaxis is unmistakable and more fatal and, assuming a similar situation in the US, the recent FDA decision regarding nasally administered noradrenaline for anaphylaxis in the light of their unseemly haste over Covid-19 vaccinations is hard to understand. Could political and financial pressured be involved? Surely not!
Featured Video

Iran War Is Accelerating the End of US Dominance
or go to
Aletho News Archives – Video-Images
From the Archives
Why the UK, the EU and the US Gang-Up on Russia
By James Petras :: 03.20.2018
Introduction: For the greater part of a decade the US, the UK and the EU have been carrying out a campaign to undermine and overthrow the Russia government and in particular to oust President Putin. Fundamental issues are at stake including the real possibility of a nuclear war.
The most recent western propaganda campaign and one of the most virulent is the charge launched by the UK regime of Prime Minister Theresa May. The Brits have claimed that Russian secret agents conspired to poison a former Russian double-agent and his daughter in England, threatening the sovereignty and safety of the British people. No evidence has ever been presented. Instead the UK expelled Russian diplomats and demands harsher sanctions, to increase tensions. The UK and its US and EU patrons are moving toward a break in relations and a military build-up.
A number of fundamental questions arise regarding the origins and growing intensity of this anti-Russian animus.
Why do the Western regimes now feel Russia is a greater threat then in the past? Do they believe Russia is more vulnerable to Western threats or attacks? Why do the Western military leaders seek to undermine Russia’s defenses? Do the US economic elites believe it is possible to provoke an economic crisis and the demise of President Putin’s government? What is the strategic goal of Western policymakers? Why has the UK regime taken the lead in the anti-Russian crusade via the fake toxin accusations at this time?
This paper is directed at providing key elements to address these questions. … continue
Blog Roll
 Aletho News
Aletho News- Trump White House plagiarized Iran war manifesto from Israel-aligned think tank
- Western media whitewashes Israel’s attempted murder of British journalist in Lebanon
- Israel-linked arms facility set ablaze in EU
- IRAN: The Three Islands Unmaking American Unipolar Sea Power in the Persian Gulf
- Trump’s dispatch of Marine Expeditionary Unit signals desperation for any symbolic success
- Iran Proposes Creating Middle East Security Structure With All Regional States – President
- Iran’s weapons industry still churning out missiles despite war: IRGC
- Washington approves billions in new arms sales to Gulf states as concerns grow over stocks of air defenses
- Iran War: Pentagon’s $200B Budget Could Run Out in Just Five Months
- Seven US allies endorse Hormuz ‘coalition,’ offer ‘no commitment’ for military action
- If Americans Knew
- Meet the former fashion blogger and shady doctor behind the ‘30,000 dead’ Iran psy-op
- Vatican Secretary of State to Trump, Israel: End the war as soon as possible
- The Majority of Americans Believe War Against Iran Benefits Israel More Than US
- Efforts to shut down pro-Palestinian speech face series of setbacks in court
- What has Israel done to my brother?
- Eid without worship in Al Aqsa – Not a ceasefire Day 161
- Trump: Israel attacked Iranian gas field without US knowledge. No more such attacks!
- America’s friends must help extricate it from an unlawful war
- Democratic Nat’l Committee’s Kneejerk Backing of Israel Is Political and Moral Failure
- Major revelations from former counterterrorism chief Joe Kent’s extensive interview
- No Tricks Zone
- Energy Expert: Germany’s Nuclear Phaseout Was A “500 Billion Euro Mistake”
- New Research: South Australia’s Mid-Holocene Sea Surface Temperatures Were 4°C Warmer Than Today
- Storing Green Energy To Last Germany 10 Days Would Require A 60-Million Tonne Battery
- New Studies: UK Sea Levels Were 4 Meters Higher Than Today During The Mid-Holocene
- Destructive Green New Deal: German Energy And Metal Group Warns Of Drastic Crisis
- New Study Documents A 20-Year Pause In Arctic Sea Ice Decline – Driven By Internal Variability
- Wake-up Call: Survey Shows Majority Of Germans Now Favor Postponing Climate Targets!
- Televised! Leading German Political Candidate Tells Schoolchildren CO2 Makes Sun Hotter!
- New Study: A Century Warming Of 1.1°C Is ‘Commonplace’ And ‘Not Unusual’ During This Interglacial
- New Study: ‘Internal Noise’ And Volcanic Forcing Can Trigger 10-15°C Warming Within Decades
