On August 31, 2023, roughly two weeks before the latest COVID booster recommendation by FDA/CDC/ACIP, independent researchers published an online, open-access analysis announcing “… it is possible to distinguish, by tryptic digestion, followed by mass spectrometry analysis, synthetic Spike proteins originated from the translation of the mRNA vaccines from natural Spike circulating in biological fluids.”
In other words, the researchers devised a method to determine how long the synthetic spike protein created by mRNA vaccines was present in the human body of vaccinated individuals.
This was a big deal. Their approach represents the first proteomic detection of recombinant Spike in vaccinated subjects. How long did they find it lasted?
They write, “The specific PP-Spike fragment was found in 50% of the biological samples analyzed, and its presence was independent of the SARS CoV-2 IgG antibody titer. The minimum and maximum time at which PP-Spike was detected after vaccination was 69 and 187 days, respectively.” Below is a chart from their study comparing the detection of the spike protein in the body of vaccinated and “after infection non vaccinated.”
The FDA and CDC’s Advisory Committee on Immunization Practices (ACIP) didn’t care and approved the new COVID booster anyway calling for yearly boosters ‘like the flu shot.’
The mRNA vaccine technology used against a circulating coronavirus is a new, never-before-used approach and method. Proper safety testing and an understanding of what happens when you repeatedly inject billions is unknown. Why the approval? Well because it was an emergency we were told… we just didn’t have time. You understand right?
Yet, with the newly updated booster announced by the CDC for 6 months and older, with no exceptions we are still aggressively injecting. Why? America is the outlier with its cavalier approach to experimenting on its population as new warnings appear almost weekly from this injectable tech platform.
The CDC states mRNA vaccines use mRNA created in a laboratory to teach our cells how to make a protein that triggers an immune response inside our bodies. The Covid vaccine’s one and only purpose is to create that spike protein. It has only one job.
Do you think regulators or the pharmaceutical companies making the shots cared to understand what else that spike did after it was created or how long it persisted?
Pfizer’s Nonclinical overview submitted to FDA’s Center for Biologics Evaluation and Research, a document which doctors had to sue the agency in court to obtain, states:
“The protein encoded by the RNA in BNT162b2 is expected to be proteolytically degraded like other endogenous proteins… Therefore, no RNA or protein metabolism or excretion studies will be conducted.”
In other words, we aren’t going to bother looking because one can’t find what one doesn’t search for.
Two years later, public assertions like the one from The Infectious Disease Society of America still regularly repeated estimates that the spike proteins generated by COVID-19 vaccines last up to a few weeks.
“I’ve asked this before and I just don’t have a clear idea about how long the spike protein the messenger RNA in our bodies produce… how long has it been detected in patient serum or tissues or even in animal studies? Do you know how long it may persist in blood or serum or tissues?”
To this question, Rituparna Das, Moderna’s Therapeutic Area Head of Respiratory Vaccines and the company’s ACIP lead answered:
“The spike protein, ah, availability I believe is on the order of days, but, like less than a week. But I will confirm that with our tox [toxicology] folks as well.”
No one knew for sure, not even the injectable product’s manufacturer, and more importantly, no one cared to know.
Yet the spike continued to turn up as the culprit in more pathogenic insults to humanity. In 2023, researchers reporting circulating spike protein detected in post–COVID-19 mRNA vaccines myocarditis stated:
“A notable finding was that markedly elevated levels of full-length spike protein, unbound by antibodies, were detected in the plasma of individuals with post vaccine myocarditis, whereas no free spike was detected in asymptomatic vaccinated control subjects”
The CDC/ACIP response was that the benefits outweigh the risks… simplistic, insulting, and lacking transparency talking down to the people being targeted by this novel shot.
The authors of the August 2023 suggested that the spike protein may be integrating into the human cells. A similar, and in ways more detailed, warning was just given by well-respected cancer genomics researcher at the University of South Carolina Phillip Buckhaults, Ph.D. during his recent testimony in front of the South Carolina Senate Medical Affairs Committee which he stated, among other things, that:
The Pfizer mRNA vaccine is contaminated with the plasmid DNA vector that was used as the template for in vitro transcription reaction.
This DNA could cause rare but serious side effects like death from cardiac arrest.
The DNA can and likely will integrate into the genomes of transfected cells.
There is a very real hazard for genome modification of long-lived somatic cells, which could cause sustained autoimmune attacks towards that tissue.
There is also a theoretical risk of future cancer, depending on the piece of DNA and site of integration.
The CDC’s website states the following outdated and scientifically lazy explanation, at best, of what happens to human DNA after being injected with COVID-19 shots.
According to Professor Buckhaults, this is just not true… at all.
Two further points raised by Professor Buckhaults were first, that the plasmid DNA contamination was not present in the material used in Pfizer’s initial vaccine trials for regulatory approval. It was only after approval and rapid scale-up of manufacturing did the company used questionable, scientifically reckless techniques which led to the contamination. Second, Professor Buckhalut’s lab, a world leader in this type of research, estimates that each vaccination contains about 200 billion pieces of plasmid DNA encapsulated in the lipid nanoparticle.
We know it is now a basic technique to find the synthetic spike in vaccinated individuals. Perhaps even more troubling, it’s basic genetic research to find out if the plasmid DNA is integrating into and forever changing the DNA/genetics of vaccinated individuals yet health agencies and labs just don’t seem to want to look.
With the CDC clearly not willing to do even the most basic steps to regain the public trust lost, as new director Cohen claimed was her main goal, the public must back away further from an apparently rogue government body. As prominent scientists and doctors denounce the agency and its products, we have hit breakaway speeds into historically uncharted territory as public health agencies, once a fixture running in the background of America, have become a cyclic, menacing threat with each new booster rollout campaign.
Uttering words that would have seen you excommunicated from ‘good’ society, ostracized to an island of racists, bigots, and vaccine-deniers; Florida Surgeon General Dr. Joseph Ladapo said in a statement that the vaccines “are not backed by clinical evidence, but blind faith alone with ZERO regard for widespread immunity.”
In guidance (pdf) to patients and doctors, the Florida Department of Health added:
“Based on the high rate of global immunity and currently available data, the state surgeon general recommends against the COVID-19 booster for individuals under 65. Individuals 65 and older should discuss this information with their health care provider, including potential concerns outlined in this guidance.”
This directly contradicts guidance from The White House (everyone get up to date) and the CDC and FDA (endorsing the new jabs for anyone over 6 months old):
“We continue to live in a world where the CDC and the [Food and Drug Administration], when it comes to COVID at least, are just beating their own path in a direction that’s inexplicable in terms of thinking about data and in thinking about common sense,” Ladapo said.
And three years into this flu season, Ladalpo highlights ‘herd immunity’ among most of America:
“With the amount of immunity that’s in the community – with virtually every walking human being having some degree of immunity, and with the questions we have about safety and about effectiveness, especially about safety, my judgment is that it’s not a good decision for young people and for people who are not at high risk at this point in the pandemic,” he said.
Florida Governor DeSantis agreed:
“I will not stand by and let the FDA and CDC use healthy Floridians as guinea pigs for new booster shots that have not been proven to be safe or effective,”
In March, the CDC and FDA sent a letter to Ladapo, warning that he was fueling vaccine hesitancy and harming Florida’s seniors.
Ladalpo is not alone in his scepticism.
“Pushing a new COVID vaccine without human-outcomes data makes a mockery of the scientific method and our regulatory process,” Drs. Marty Makary and Tracy Beth Hoeg said in an op-ed.
“If public-health officials don’t want a repeat disappointing turnout of Americans who get the COVID booster shot, they should require a proper clinical trial to show the American people the benefit,” they added.
Just 17 percent of Americans received one of the bivalent doses, which were made available in the fall of 2022. The new vaccines replaced the bivalents.
“The CDC is advising the children get these boosters when there’s no evidence that children receive any benefit and clear evidence that they receive harm,” Dr. Robert Malone, who helped invent the messenger RNA (mRNA) technology the Pfizer and Moderna vaccines use, said on EpochTV’s “Crossroads.”
Risks include myocarditis, a form of heart inflammation that can lead to sudden death.
And cue the mainstream media ‘blood on their hands… science-denying’ headlines.
The Fifth Circuit Court of Appeals reversed a lower court’s ruling that “sovereign immunity” protects the Food and Drug Administration (FDA) from any wrongdoing or harm in telling the public to stop taking ivermectin, a safe, well-studied, and proven drug for the prevention and treatment of COVID-19.
In their opinion, Judges Clement, Elrod, and Willett state, “FDA argues that the Twitter posts are ‘informational statements’ that cannot qualify as rules because they ‘do not ‘direct’ consumers, or anyone else, to do or refrain from doing anything.’ We are not convinced.”
“We are very pleased with this development and extremely proud of our colleagues for taking a stand against a government health agency that is clearly overstepping its authority,” said Pierre Kory, M.D., M.P.A., president and chief medical officer of the FLCCC. “The FDA’s campaign against ivermectin continues to be used as an excuse by hospitals to deny access to a lifesaving treatment and weaponized by medical boards to threaten the licenses of doctors who stray from the mainstream to prescribe a drug that has been proven in controlled trials to safely treat hundreds of thousands of patients around the world.”
The lawsuit, Apter et al v. Dep’t. of Health and Human Services et al, was brought by Robert Apter, MD, Mary Talley Bowden, MD, and FLCCC co-founder, Paul E. Marik, MD, and first filed in the US District Court on June 2, 2022. It stated that the FDA acted outside of its authority and illegally interfered with the doctors’ ability to practice medicine with an aggressive effort to stop the prescribing of ivermectin for the prevention and treatment of COVID-19.
The case was later dismissed by the court citing that the FDA had “sovereign immunity,” giving the agency absolute protection from any wrongdoing or harm in directing the public, including health professionals and patients, to not use ivermectin, a drug that has received full FDA approval for human use. Earlier this year, Apter et al filed an appeal in the US Court of Appeals for the Fifth Circuit requesting the Court reverse the lower court’s dismissal of the lawsuit.
The Court’s reversal was issued yesterday with the ruling, which said “FDA is not a physician. It has authority to inform, announce, and apprise—but not to endorse, denounce, or advise. The Doctors have plausibly alleged that FDA’s Posts fell on the wrong side of the line between telling about and telling to.”
The ruling goes on to say the “FDA can inform, but it has identified no authority allowing it to recommend consumers ‘stop’ taking medicine.” And finally, “Even tweet-sized doses of personalized medical advice are beyond FDA’s statutory authority.”
“The work of the legal team at Boyden Gray has been nothing short of superb,” Kory added. “We are very fortunate to have them on the side of our doctors in this case.”
The Fifth Circuit Court’s ruling can be found here:
The FLCCC filed its amicus brief in support of the lawsuit in February of this year. A copy of the brief can be found here.
About the Front Line COVID-19 Critical Care Alliance
The FLCCC Alliance was organized in March 2020 by a group of highly published, world-renowned critical care physicians and scholars with the academic support of allied physicians from around the world. FLCCC’s goal is to research and develop life-saving protocols for the prevention and treatment of COVID-19 in all stages of illness including the I-RECOVER protocols for “Long COVID” and Post Vaccine Syndrome. For more information: www.FLCCC.net
For those of us who started pulling uncomfortable threads of ‘hang on a minute, that doesn’t seem quite right’ and kept going, the world is now a strange landscape. When it comes to our health, many of us have had life-long assumptions smashed to smithereens.
The last few years have been a brutal introduction to the harsh realities of the medical industrial complex. Its recent behaviour has completely shattered public trust. Overnight, medical ethics including bodily autonomy and informed consent were tossed out. The dying were unable to see loved ones, blanket DNRs were applied and people were subjected to forced procedures (e.g. PCR testing) or were discriminated against based on their covid vaccination status. Most went along with these inhumane diktats that came from ‘on high’. Is it any wonder that many people would now be reluctant to enter a hospital even where there was a genuine need? There has to be a balance where people regain the confidence to seek care if genuinely needed. Throwing the baby out with the bath water will not lead to fewer excess deaths. Patients rightly want the best available care but in the current system of protocolised medicine, that pathway is far from guaranteed.
What is striking, when spending time amongst those still firmly planted behind the Overton window, is the notion that for any ailment – particularly with advancing age – the answer is always pharmaceutical or surgical interventions rather than lifestyle changes. This is clearly superb for Big Pharma. Less certain, is the benefit for the end user. Blanket approvals of the novel, mRNA injections across all age groups – in spite of total lack of long-term safety data – showed us clearly, in real-time, how money and politics corrupts health regulators. This renders the current system totally defunct. This article in The Epoch Timesreports how 65% of drug recommendations by the FDA are approved based on a single study. In the meantime doctors who want to promote lifestyle changes as first line treatment ahead of more dangerous interventions, find themselves increasingly working in a system opposed to them.
Trying to make an informed assessment of risks and benefits becomes almost impossible, once you realise how untrustworthy scientific literature has become. Or perhaps always was. ‘But it says so in the Lancet…’ Hopefully, many have now realised how empty this sentence sounds. Research outcomes are heavily influenced by the desires of those funding them, who just happen to be the ones manufacturing the drugs. This meme, whilst humorous, is worryingly accurate:
From statins to HRT, to proven-to-be-ineffective surgeries, to antidepressants, there is almost zero discussion in modern ‘medicine’ of preventative measures that do not benefit Big Pharma. Diet, exercise, breathing techniques, stress management, ensuring proper balance of micronutrients, enquiring about emotional and relationship causes of ill health. These should be the basic lines of enquiry for any competent physician, long before offering various magic bullets following the luxurious seven minute consultation via Zoom. GPs offering this paltry level of service are complicit in ensuring their own redundancy within a few short years. AI would serve just as well, the human factor having been almost entirely scrubbed out.
Most people with elderly parents know that they often need special plastic containers to house the numerous medications that they take on a daily basis. Often many of these drugs were added in sequence to deal with side effects (or more correctly, ‘effects’) of the medications that were introduced first. This close-to-the-bone satire that has been circulating online really sums it up:
“I took ASPIRIN for the headache caused by the ZYRTEC™ for the hay fever I got from the RELENZA™ for the upset stomach and flu-like symptoms caused by the VIAGRA™ for the erectile dysfunction from the PROPECIA™ for the hair loss caused by the RITALIN™ for my short attention span caused by the SCOPODERM TTS™ for the motion sickness that I got from the LOMOTIL™ for the diarrhoea caused by the XENICAL™ for the weight gain caused by the PAXIL™ for the anxiety that I got from the ZOCOR™ that I’m taking for my high cholesterol, because a good diet and exercise is just too much trouble.”
There seems to be the prevailing belief that human beings can only stay alive with constant interference from the medical profession. But what if the entire system keeps you sick, in order to retain you as a loyal customer to The Firm? What if much of the so-called ‘safety’ data are in fact just as flawed as those used to push the covid ‘vaccines’ in 2020? For those of us who became curious (suspicious?), we felt it might be worth having a retrospective look at other trends in medical diagnostics and treatments.
As it turns out, the more you look, the more you find…
Doing more harm than good?
A deep dive into questionable drugs and surgical practices that are built into standard protocols here in the UK would fill a book. Maybe several books. That is beyond the scope of this article, however here are a few ‘top picks’ to get started:
Statins: This has been allowed on occasion to seep into mainstream consciousness, but it is still worth reading the book written by Dr Malcolm Kendrick, entitled The Great Cholesterol Con. There is simply no good evidence for the widespread use of statins. The notion that they don’t have harmful effects is also nonsense. A 17-year study on the elderly showed that low serum cholesterol was associated with increased frailty, accelerated mental decline, and early death. This should get any sensible clinician asking questions about what effects deliberately lowering it might have on long-term health. During ward rounds one senior consultant says to his juniors, ‘let’s stop this wonder drug’, scoring it off the prescription list. The juniors ask why he calls it that, to which he replies ‘I wonder what use it is!’
SSRIs: a growing number of studies show they are less effective than thought. Has this resulted in a decline in prescriptions? Of course not. In fact psychiatry in general is an area that many medics describe as barbaric. The links between gut and brain health are now widely accepted and yet almost no money goes into mainstream R&D in this area. Cures are simply not as profitable as life-long customers.
HRT: There is evidence for an increased risk of blood clots and stroke as well as an increased risk of breast and ovarian cancer in women using HRT. In researching this article and questioning women who had recently started on these drugs, most had not been adequately informed as to any associated risks. Many had no idea what kind of HRT they were even taking. This shows the level of blind faith people still have in a system that, judging by the history of criminal fines paid out, does not have a very good track record, to say the least.
Angioplasty: For decades, we were led to believe that angioplasties are an effective treatment for not only angina (chest pain) but also served as protection from a heart attack. Now, the evidence seems to point to the procedure being ‘useless’ or even worse than useless. Lifestyle and diet changes are more effective and have the capacity to reverse the progression of coronary heart disease. This hardly seems like rocket science but somehow has taken decades to ‘realise’.
Childhood Vaccinations: or as we like to call them, The Sacred Cows. Daring to even utter the words that these interventions may carry risks as well as benefits, or pointing out that there are no long-term safety studies using a genuine placebo, seems to create an allergic reaction in even the most sceptical of folks. However, at HART the adage ‘everything is back on the table’ is one we hold dear. Many people who now have the appetite to question The Science™ are quietly murmuring the name of the book Turtles All the Way Down. Perhaps it is worth reading, just to know what the alternative view point is. We do not need protection from theories. We need them to be aired and debated, so we can reach full and informed decisions. No topic should be out of bounds, including this one. Perhaps especially this one.
This list could go on ad infinitum, but instead we will end with a list of suggestions given by various practising medics when asked about drugs or interventions that they question. Once again, we point out that people are individuals. Protocols are not good for individuals. Do your research, take responsibility and remember that The Experts may have their hands tied firmly behind their back by The Money.
List of drugs with questionable efficacy/safety from currently prescribing doctors:
Benzodiazepines and other so-called ‘Z’ drugs (zopiclone, eszopiclone, zaleplon and zolpidem): habit forming with evidence of severe side effects such as dementia, infections, respiratory disease exacerbation, pancreatitis, and cancer. They cause severe withdrawal symptoms;
Gliflozins: the new ‘wonder drugs’ used in diabetes and heart failure. Concerns about kidney damage and they come with no long-term safety profile;
GLP-1 agonist injectables: originally marketed for diabetes, now being sold as weight loss drugs. Concerns over risks of thyroid and pancreatic cancer and can cause pancreatitis;
Psychiatric drugs in general. Recommended reading: Toxic Psychiatry by Peter Breggin;
Antivirals: For example Tamiflu, which is now labelled a ‘fiasco’. The drug caused severe side effects such as hallucinations, self-injury, abnormal behaviour and renal impairment;
Proton Pump Inhibitors (PPIs):evidence of major side effects with long-term use, including dementia, chronic kidney disease and increased cancer risk;
Beta Blockers:evidence that long-term use is not associated with improved cardiovascular outcomes, with considerable side effects, such as depression and fatigue;
Anti-arthritis drugs: Overuse of NSAIDs in particular can cause bleeding, heart attack, stroke, and renal damage.
To quote Aldous Huxley: ‘Medical science is making such remarkable progress that soon none of us will be well’.
The gist of a current court case that you’ve likely never heard of, is that three heroic doctors are suing the FDA about the loss of their jobs, about their careers being derailed, about the loss of their reputation — all because their professional, scientific opinion as to what was in the best interest of their patients, was different than the political agenda of the FDA. (Here is a bit of background.)
What is at stake here could not be more significant, and it applies across the board to EVERY federal agency. The question is: do federal agencies have the unsupervised right to replace Science with political science? Put another way: can they act dishonestly, incompetently, etc. with essentially no meaningful consequences?
Here is the doctors’ Complaint. Although it was filed a year ago, it is now being appealed this week — and some fascinating audio clips have emerged. There are three judges on a panel, asking the attorney representing the FDA some probing questions.
Five of these short audio clips (3-5 minutes each) are posted here. (The recording of the full proceeding is here.)
IMO some of the key takeaway revelations (so far) are:
1 – The FDA seems to claim that their published warnings are little more than offhand observations. For example, their slamming of Ivermectin was evidently just casual commentary (what the FDA calls “informational”).
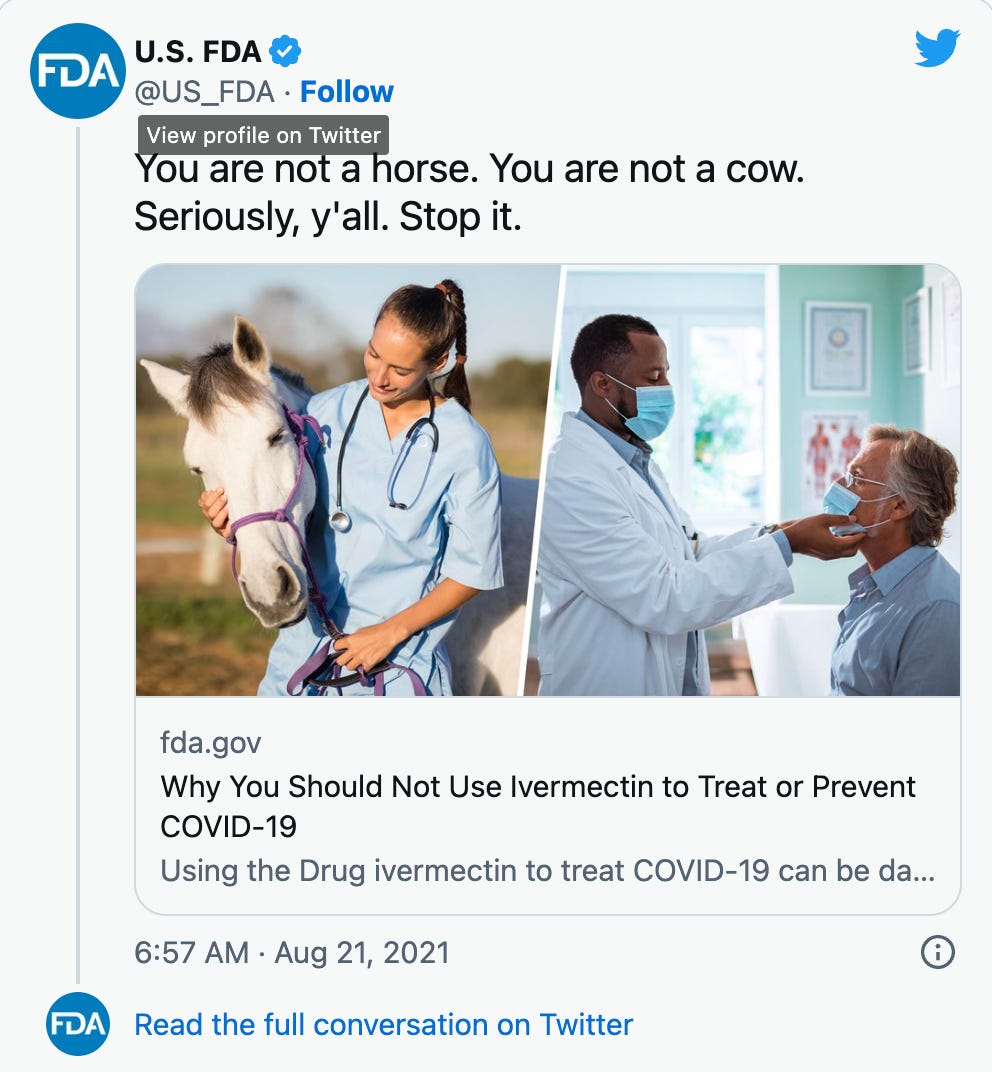
Note the title here, on this FDA page which is STILL up! It says “Why You Should Not Use Ivermectin to Treat or Prevent COVID-19”.
Note 1: This is a deceptive headline because that article is mostly saying: a) citizens should not self-medicate, and b) using any veterinary medications can be dangerous. Both of these are legitimate concerns. So, if the FDA was honestly trying to benefit the public their heading should be: “Why You Should Not Self-Medicate Using Veterinary-Grade Ivermectin to Treat or Prevent COVID-19”. BIG DIFFERENCE!
Note 2: This FDA page has changed quite a bit over time. Here is the 2021 version.
Note 3: The current page makes outright false statements like: “Ivermectin has not been shown to be safe or effective for these indications.” I’m one of the few people who has taken the time to put together a spreadsheet of ALL the studies on ALL the major COVID early treatment therapies: see it here.
There have now been 99 Ivermectin scientific studies, and the overall early treatment effectiveness is 62%. IVM’s extensive safety record is extraordinary, with adverse effects (e.g., see here) in the ballpark of only one in a million usages!
Now, also on my spreadsheet, compare what the FDA has approved for early treatment of COVID-19 therapy: Paxlovid = 32% effective with these adverse safety issues, and Molnupiravir = 16% effective with these problematic safety issues!
Despite these LARGE benefits of Ivermectin in effectivenessandsafety, the FDA continues to say that “Ivermectin has not been shown to be safe or effective” for early treatment of COVID-19. This is stunningly inaccurate.
Note 4: Even though the FDA now has access to 99 Ivermectin studies, their statement against Ivermectin is stronger now than when the page originally appeared in 2021! IMO this is what happens when a federal agency feels that there is no meaningful oversight, so effectively they can say anything they want.
2 – The FDA says that Courts have no business in reviewing anything they say or do!
Considering the above facts in #1, it’s obvious why this would be their self-serving position. Listen carefully to the second short audio clip, where the FDA’s attorney appears to say that the FDA’s communication to the public can be knowingly false, dishonest, etc. with no oversight or consequences — even when deaths result!
Regretfully, to date, the courts have played along with this game of charades. For example, the Chevron case is frequently cited by non-aggressive attorneys to say that courts will stay out of determining whether FDA processes, documents, and claims are legal, accurate, honest, warranted, etc.
However, that is an oversimplified opinion. Even the Chevron case states that the FDA’s actions must be “reasonable” — but that is rarely argued. BTW, the case we are discussing here would never have been filed if the doctors’ attorneys bought into the bogus idea that federal agencies have unlimited deference. Kudos to them that they did not accept that absurd argument!
Maybe I’m overly optimistic, but based on the judges’ questions and comments in these clips, it seems to me that this case might eventually upend Chevron. That would be EXTRAORDINARILY beneficial for US citizens, as it would apply to all national policies: from immigration to education, energy to climate change, etc.
3 – The FDA asserts that the only recourse that US citizens have about even egregious errors and deceptions by the FDA is through the “political process.” Astounding!
4 – The FDA indicated that the “political process” means that citizens need to elect a competent and attentive President, whose responsibility it is to see that the FDA acts responsibly — or else. The flip side is that when we do not have such a President, all federal agencies have a four-year time period to wreak whatever political havoc that suits them — again, across the board, and without real consequences to the guilty parties.
5 – The FDA’s attorney implied that there would be no compensation given for inaccurate or knowingly false FDA statements — including those that lead to Americans unnecessarily dying — other than an FDA person may lose their job.
6 – Based on these select audio clips, the fact that hundreds of thousands of Americans likely died needlessly due to the FDA’s COVID actions and inactions (see here), was not fully addressed. Hopefully, this will be brought up in this trial.
7 – In clip #3, the FDA attorney makes the startling claim that the FDA has the authority to give citizens medical advice! How is that possible when they know nothing of the medical history of any American citizen? Further, once they assert that right, how is a conflict resolved between what the FDA says and what a citizen’s medical provider says? That is one of the major issues in this important case.
8 – In clip #4, the FDA attorney acknowledges that doctors have lost their jobs, etc. due to their scientific conclusions on such matters as Ivermectin, and their science-based actions that they believed were in the best interest of their patients. However, the FDA attorney then stated that no losses, etc. were due to anything the FDA did. (!)
……….
Note that a lot of the bad behavior with the FDA (and CDC) would be reduced if the Medical Establishment refused to play politics and instead supported real Science for the public. Regretfully, that has not happened and the COVID-19 fiasco exposed this ugly underbelly. See my Report on the COVID failings of the Medical Establishment.
In another Report, I compared the FDA’s approval process for Remdesivir to Ivermectin. This appears to show stunning incompetence at the FDA.
I have made this point before, but it’s worth repeating. The war we are engaged in is that powerful Left-wing forces (exterior and from within) are trying to take America down. One of their primary strategies to do this is to replace Science with political science. That is what this case is about, as the FDA is specifically arguing that they have the right to scrap Science and substitute political science — with impunity!
Draw your own conclusions, but to me, this case is like a Molotov cocktail thrown into the Federal Government bureaucracy. Astoundingly, all three branches of our government are complicit with this nonsense.
Some obvious questions that need to be answered and fixed are: 1) How did Congress give pharmaceutical companies such broad protections against self-serving unscientific actions? 2) How did the Executive branch allow agencies like the FDA to be run by parties that they are supposed to regulate? 3) How did our Judicial system allow bad actor agencies to arrange to have no real legal oversight?
Considering that these failings are applicable to multiple federal agencies, is there any question why such things as COVID policies (and energy, and climate, and education, and immigration, and elections, etc., etc.) are a disaster?
Hopefully, this lawsuit will crack open the door to fixing this horrific mess…
……….
PS — What needs to be done now :
1) Competent attorneys should file friend of the court briefs to support this nationally important case. Overturning the Chevron precedent would have extraordinarily positive benefits for almost ALL US citizens.
2) Competent federal legislators should introduce a “Save America” bill (aka Agency Oversight Act). This legislation will rein in ALL federal agencies, by providing timely and meaningful oversight (plus real penalties) to them all.
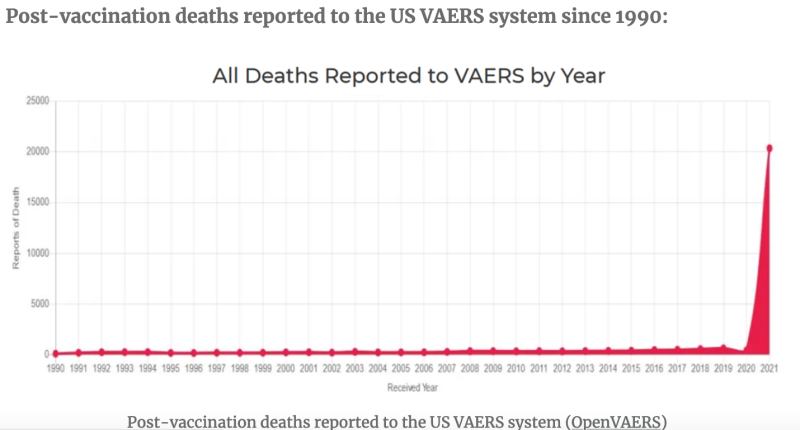
The Centers for Disease Control and Prevention (CDC) V-safe website quietly stopped collecting adverse event reports with no reason or explanation. The V-safe website simply states: “Thank you for your participation. Data collection for COVID-19 vaccines concluded on June 30, 2023.” If you go there today, V-safe directs users to the FDA’s VAERS website for adverse event reporting, even though officials continually derided VAERS as “passive” and “unverified.”
VAERS and V-safe are mutually exclusive safety collection databases operated by the FDA and CDC, respectively. VAERS is an older way of collecting safety data where one can fill out a form online, or manually, or by calling a toll-free number, whereas V-safe is a device “app” which requires online registration. Both VAERS and V-safe collect personal information, lot numbers, dates and associated information, but V-safe was an active collection system geared towards a younger app-using demographic.
Does this mean that the CDC believes that the mRNA Covid-19 injections are so safe, there is no need to monitor adverse event reports any longer? What is the argument against continued monitoring, especially since the V-safe website was already up and paid for?
While CDC’s V-safe was stealthily and abruptly turned off, refusing to accept new safety reports, to this very day the CDC continues to urge everyone ages 6 months and older to stay up to date with COVID-19 vaccines and boosters.
As a drug safety expert, I personally can’t cite another example of any agency or manufacturer halting collection of safety data. It seems even worse because mRNA technology is relatively new with long-term manifestations unknown. On top of this, both manufacturers and the FDA refuse to sharethe list of ingredients, such as lipid nanoparticles, which could affect individuals differently and take a long time to manifest clinically.
Safety Data Collection Should Never Stop:
Now, contrast that with the fact that the National Highway Traffic and Safety Administration (NHTSA) will still accept a safety report for a 30-year-old Ford Bronco II. Indeed, this is an oddly specific example, but only because I drove this exact vehicle as a family hand-me-down as a student, through my residency, fellowship, for my tenure as a Yale professor on the mean streets of New Haven and even during my years at the FDA as a medical officer /senior medical analyst.
Like mRNA shots, Bronco IIs are still available on the market and people are still using them up to this very day. My Bronco became an intermittent topic of conversation with friends and FDA colleagues. One day, I was informed by a patrolling security guard at the FDA that it was the oldest car on campus.
I didn’t know much about cars (or mRNA technology) back then, but when a fellow FDA-er informed me that my Bronco II had noteworthy safety problems and that the NHTSA still had their eye on this vehicle (rollover accidents were more common and more fatal) I addressed the problem: I got rid of the reliable relic, even though I reallyliked it. NHTSA Is still accepting safety reports three decades later.
Interestingly, the NHTSA link above on my Ford Bronco II only shows: one parts recall, one investigation and 23 complaints, and still features a button in the upper right hand corner for submitting new complaints.
Wikipedia defines an humanitarian crisis or humanitarian disaster as a: “singular event or a series of events that are threatening in terms of health, safety or well-being of a community or large group of people.” Based on VAERS and previous V-safe findings, adverse events from mRNA shots in the USA alone could be considered a humanitarian crisis.
Despite those alarming clinical findings, the CDC has concluded that collecting new safety reports is somehow no longer in the interest of America’s public health. Existing data from the V-safe site showed around 6.5 million adverse events/health impacts out of 10.1 million users, with around 2 million of those people unable to conduct normal activities of daily living or needing medical care, according to a third-party rendering of its findings. In other words, despite mRNA shots still being widely available and the CDC promoting its continued use, it’s “case closed” with regards to collecting new safety reports, under today’s federal public health administration.
Will the CDC opine on the existing data or justify its halting of collecting new safety data? To the best of my knowledge, stopping the collection of public health information doesn’t have a clinical justification or scientific precedence — especially when it comes to an actively marketed product.
In George Orwell’s 1984, characters were told by The Party to “reject the evidence of your eyes and [your] ears.” Now, the CDC isn’t even allowing that evidence to be collected for viewing (and prospective rejecting). It’s a terrible idea for any product, let alone novel mRNA technologies.
Dr. David Gortler, a 2023 Brownstone Fellow, is a pharmacologist, pharmacist, research scientist and a former member of the FDA Senior Executive Leadership Team who served as senior advisor to the FDA Commissioner on matters of: FDA regulatory affairs, drug safety and FDA science policy. He is a former Yale University and Georgetown University didactic professor of pharmacology and biotechnology, with over a decade of academic pedagogy and bench research, as part of his nearly two decades of experience in drug development. He also serves as a scholar at the Ethics and Public Policy Center.
The Epoch Timesrecently reported an astonishing statement by a U.S. government lawyer in a federal court in Texas, where the FDA is being sued by Dr. Paul Marik of Virginia, Dr. Mary Bowden of Texas, and Dr. Robert Apter of Arizona. The three plaintiffs claim the FDA illegally prohibited them from prescribing the drug to their patients. At a November 1 hearing, U.S. lawyer Isaac Belfer argued for the defendant:
The cited statements were not directives. They were not mandatory. They were recommendations. They said what parties should do. They said, for example, why you should not take ivermectin to treat COVID-19. They did not say you may not do it, you must not do it. They did not say it’s prohibited or it’s unlawful. They also did not say that doctors may not prescribe ivermectin.”
If Belfer’s assertion is true, it raises a very urgent question: On what legal grounds did hospitals all over the United States refuse to administer ivermectin to severely ill COVID-19 patients, even when patients and their family members begged for the drug to be administered?
If ivermectin was not prohibited by the FDA or any other U.S. medical authority for treating COVID-19, why did Dr. Paul Marik’s hospital prohibit him from administering the drug to his dying patients? Why was Dr. Mary Bowden reported to the Texas Medical Board for disciplinary action when she prescribed it? Why did many pharmacists fear losing their licenses if they filled ivermectin prescriptions for treating COVID-19?
All these patients asked for was to be allowed to try the drug (FDA-approved for River Blindness, Elephantiasis, and Scabies) for COVID-19. The patients and their kin gladly indemnified the hospitals and arranged to have their independent primary care doctors deliver and administer the drug. Nevertheless:
Hospital administrators absolutely refused to grant this wish.
Hospital attorneys fought tooth and nail against using ivermectin to treat COVID-19 patients, doing everything in their power to challenge patient lawsuits and appeal court orders to administer the drug.
Even when hospital doctors acknowledged that the patients were dying, they insisted it was better to let the disease take its natural course rather than allow patients to try ivermectin.
Even when patients’ families succeeded in getting a court orders to administer the drug, many hospitals still refused, even at the risk of being held in contempt of court.
Several readers have told us that our chapters covering this shameful scandal— Chapters 38: Begging for the Wonder Drug and Chapter 40: Graduating into Eternity—are horrifying beyond belief.
Now we hear U.S. government lawyers arguing in court that the FDA never prohibited using ivermectin to treat COVID-19 patients, but merely recommended not using it. This indicates that hospitals had no legal grounds for denying sick patients a drug that could have helped them. How is withholding medicine from a sick man any different from withholding a life ring from a man who has fallen overboard in high seas?
For families who watched their loved ones slip away after being denied the right to try ivermectin, U.S. attorney Isaac Belfer’s statement may be interpreted as declaring open season for lawsuits against hospital administrators and doctors.
Nevertheless, Dr. Marik and his co-plaintiffs, Robert L. Apter and Mary Talley Bowden, appealed the dismissal and are now being heard before a three-judge panel of the 5th U.S. Circuit Court of Appeals.
Once again, attorneys for the U.S. government are in the hot seat about their mendacious claims about the FDA’s directive to doctors and hospitals against prescribing or administering Ivermectin, either to outpatients or to patients dying in hospital.
Instead of acknowledging the obvious reality that the FDA did indeed DIRECT doctors and hospitals against administering Ivermectin, U.S. attorneys continue to insist that the FDA’s communiques were mere advice.
This preposterous argument not only overlooks the plain language of the FDA’s communiques, it also overlooks the salient fact that numerous doctors (like Paul Marik) were fired from their jobs for administering ivermectin to their dying patients, and the fact that many State Medical Boards revoked doctors’ licenses for doing the same. If these punitive actions taken against doctors were NOT based on the FDA’s directives, on what grounds were they taken?
As was just reported by Just the News columnist Greg Piper:
The 5th Circuit panel seemed skeptical of Civil Division Appellate Attorney Ashley Honold’s argument that the FDA’s “informational statements” against ivermectin, including its conflation of human and animal dosages, were “merely quips” about reported problems after “self-medicating” rather than “prohibit[ing] anyone” from using ivermectin.
Judge Jennifer Walker Elrod cited the phrase “Stop it” in the agency’s viral “You are not a horse” post on X, then known as Twitter. “If you were in English class, they would say that was a command. … That is different than ‘we’re providing helpful information,'” she told Honold.
Readers of this Substack will probably agree with my sentiment that enough is enough of lying and obfuscating U.S. government agency officials and their mercenary lawyers. It’s time for the grown-up, reasonable citizenry of this country to join Marik, Bowden, et al. in suing the pants off the FDA and other U.S. agencies against whom there is a preponderance of evidence that they have unlawfully interfered with the doctor-patient relationship and committed negligent homicide, fraud, and concealment.
Cry havoc and let slip the plaintiffs’ attorneys! Sue the FDA; sue doctors and hospital administrators; and sue the medical boards. Let them pay for the damages they have inflicted on the families of patients who were denied ivermectin until their last breaths. Let them pay for the massive damage and distress they have caused for courageous doctors like Paul Marik and his colleagues who tried to help their patients.
The U.S. House of Representatives Select Subcommittee on the Coronavirus Pandemic wants to know more about plans by the Centers for Disease Control and Prevention (CDC) to recommend annual COVID-19 vaccines.
During a July interview with Spectrum News, CDC Director Mandy Cohen said she “anticipate[s] that COVID will become similar to flu shots, where … you get your annual flu shot and you get your annual COVID shot.”
“It is unclear if the science supports such a recommendation. If this anticipated CDC recommendation occurs, it will mark a significant change in federal policy and guidance regarding COVID-19 vaccines and the way in which they are utilized.”
Wenstrup requested all documents and communications about any annual — “or any other time-based iteration” — recommendation for COVID-19 booster shots, including correspondence between or among the CDC, U.S. Department of Health and Human Services (also under the subcommittee’s investigation), the White House, the CDC Foundation, CDC contractors and any other CDC stakeholders.
The committee of 21 independent advisers in June voted unanimously that any new vaccine should protect against just one strain of the virus — a departure from the available bivalent vaccines — and should target one of the three Omicron subvariants currently circulating, including XBB.1.5.
The XBB.1.5 variant spread globally in the first quarter of 2023, reaching dominance in North America, and other parts of the world by April, according to the FDA’s briefing document for the June meeting.
Dr. Mark Sawyer, professor of clinical pediatrics at the University of California, San Diego, told CNBC that describing COVID-19 as seasonal “could be problematic” because “we really don’t know what the COVID season is.”
“COVID-19 respiratory illness is now like a mild head cold. There is no seasonal pattern. The COVID-19 vaccines have failed to stop transmission or protect against hospitalization and death.
“The products on the market have theoretical efficacy of less than six months. Annual COVID-19 shots have no clinical indication, medical necessity, are not durable for 12 months and have never been tested for use on a yearly schedule.
“On Dec. 7, 2022 in a U.S. Senate panel on vaccines, I called for all COVID-19 vaccines to be removed from the market because they are not safe for human use. There has been no objection to that testimony from public health officials.”
NBC News reported that Dr. Peter Marks, the FDA’s top vaccine regulator, acknowledged during an FDA advisory committee meeting in January that “simplifying the COVID-19 vaccine schedule to be exactly like the flu may not be possible.”
Pfizer hopes otherwise. The drug company’s chief scientific officer, Dr. Mikael Dolsten, thinks an annual COVID-19 vaccine would improve vaccine sentiment, telling CNBC the public grew dissatisfied with mandates during the earlier stages of the pandemic.
He said:
“Unfortunately some people see vaccines as part of that [the mandates].
“I think of it like the introduction of seat belts for cars. People didn’t want to wear them at first, but over time they realized how much seat belts protect them. Now everyone uses them today. That’s kind of how the vaccine story needs to be reimagined.”
An annual schedule, Dolsten added, may help people view COVID-19 shots as another “very natural part” of protecting their health.
CDC director ‘very worried about parents not vaccinating kids’
In addition to the ambiguity surrounding COVID-19 vaccine scheduling, there is no consensus among medical experts on which patients would be recommended for an annual jab.
Dr. Paul Offit, a vaccine scientist, professor of pediatrics in the Division of Infectious Diseases at the Children’s Hospital of Philadelphia and a member of VRBPAC, took issue with not only the annual model but also with administering COVID-19 vaccines to low-risk groups.
“If the goal of the vaccine is the stated goal, which is protection against severe disease, do you really need a yearly vaccine for otherwise healthy people less than 75? I mean, is this the flu model? Because I would argue it shouldn’t be.”
Health advocacy groups and doctors argue against authorizing mRNA shots in young children and babies. As of July 28 — when data were last updated in the Vaccine Adverse Event Reporting System (VAERS) — there were 6,591 reports of adverse events following COVID-19 vaccination in children under age 6.
Cohen said she is “very worried about parents not vaccinating kids,” telling Spectrum News, “There’s plenty of other things that are hard as parents that we can’t do. This is one we can do to protect our kids.”
McCullough described Cohen as “fully entrenched in the bio-pharmaceutical complex” and “on the wrong side of every pandemic public health intervention.”
Jeffrey A. Tucker, founder and president of the Brownstone Institute, said Cohen’s career has been punctuated by “heartbreaking fear-mongering, pseudo-science, and propaganda,” adding that “she passed with flying colors all three tests of compliance: closures, masking, and vaccine mandates.”
Reduced trust in vaccines and the CDC concerns Cohen, who plans to rehabilitate that trust by focusing on “transparency, execution and building relationships with the public, health leaders and politicians.”
A survey by the Harvard T.H. Chan School of Public Health published in the journal Health Affairs found that roughly a quarter of Americans have little to no trust in the CDC for health information, including 10% who do not trust the agency at all.
The CDC currently recommends the primary series of mRNA shots, or the first two doses of the updated vaccine be given weeks apart, followed months later by a booster shot. The FDA updated its guidance for these shots in August 2022 to contain a bivalent formulation targeting the original viral strain plus the BA.4 and BA.5 Omicron subvariants.
Everyone seems to know someone taking Ozempic these days. But, it’s not all roses for the wonder weight loss drug, with serious side effects including suicidal ideations and stomach paralysis. Jefferey Jaxen reports.
A Louisiana woman who sued the manufacturers of popular diabetes and weight loss drugs Ozempic and Mounjaro is alleging the drugmakers failed to warn the public about the risk of severe gastrointestinal problems.
Jaclyn Bjorklund, 44, who on Wednesday filed the 26-page lawsuit in U.S. District Court for the Western District of Louisiana Lake Charles Division, claims she was “severely injured” after taking the two medications.
According to the complaint, Novo Nordisk and Eli Lilly, the manufacturers of Ozempic and Mounjaro, respectively, “downplayed the severity of the gastrointestinal events,” such as gastroparesis and gastroenteritis, caused by the drugs.
Gastroparesis, a disorder that “slows or stops the movement of food from your stomach to your small intestine, even though there is no blockage in the stomach or intestines” is frequently caused by diabetes. Narcotics and antidepressants also are linked to gastroparesis.
According to NBC News, Bjorklund is the first person to come forward alleging that the drugs in question cause gastrointestinal injuries.
The two drugs are part of a new category of medicines known as glucagon-like peptide-1, or GLP-1, receptor agonists. They are intended to help people with Type 2 diabetes manage their blood sugar levels. However, the medications are also commonly prescribed off-label for weight loss.
According to CBS News, “These drugs were originally developed to treat patients with Type 2 diabetes as they produce insulin and lower blood sugar. They also release a hormone that slows down digestion and keeps food in a patient’s stomach longer.”
According to CNN, “The lawsuit is seeking compensatory and punitive damages for past and future pain and suffering Bjorklund will have including health care costs and medical monitoring as well as her attorney’s fees and court costs.”
Attorneys Paul Pennock and Jonathan Sedgh, of Orlando-based Morgan & Morgan, said during a press conference, that the basis of the lawsuit is “a failure to warn,” CBS News reported. Pennock told the press:
“It is our opinion that these drugs are causing these problems. We think that the evidence is sufficient for us to be able to prove it or we would not have filed the case, and we intend to file many more in the coming days and weeks.
“[Bjorklund’s] problems have been so severe that she’s been to the emergency room multiple times, including last weekend. She’s actually even thrown up so violently that she’s lost teeth.”
This is the first lawsuit alleging the two drugs caused gastrointestinal injuries, however, lawyers representing Bjorklund said hundreds more similar lawsuits are ready to be filed by victims across the U.S.
Ozempic recently was linked to a range of other health issues, including kidney disorders and causing suicidal thoughts.
Plaintiff suffered from ‘severe gastrointestinal events’
The lawsuit states that Bjorklund, who was diagnosed with Type 2 diabetes in 2017, was prescribed Ozempic and took the drug for more than a year before switching to Mounjaro.
During this period, she experienced “severe gastrointestinal events,” including severe vomiting — which also led to the loss of teeth, gastrointestinal burning and stomach pain. As a result, the complaint states, Bjorklund “sustained severe and permanent personal injuries, pain, suffering, and emotional distress, and incurred medical expenses.”
The lawsuit also alleges the two companies “knew of the association between the use of GLP-1 receptor agonists and the risk of developing severe gastrointestinal issues, including gastroparesis and gastroenteritis.”
The companies’ “failure to disclose information that they possessed regarding the association between the use of GLP-1 receptor agonists and the risk of developing severe gastrointestinal issues, including gastroparesis and gastroenteritis, rendered the warnings for this medication inadequate,” the lawsuit adds.
According to The Hill, “While the labels for both medications note that they delay gastric emptying and can cause a variety of stomach problems — including nausea, vomiting, diarrhea, abdominal pain and constipation — they do not explicitly warn of gastroparesis as a risk.”
CNN reported that “Ozempic’sprescribing information says the most common adverse events related to the drug are nausea, vomiting, diarrhea, abdominal pain and constipation. Under a section on drug interactions, it says that Ozempic delays gastric emptying, which may impact absorption of oral medications.”
Similarly, “Mounjaro’sprescribing information also says nausea, diarrhea, decreased appetite, vomiting, constipation, dyspepsia, and abdominal pain are the most common adverse events, and that Mounjaro delays gastric emptying, which may impact medication absorption.”
However, due to the lack of an explicit warning regarding gastroparesis, the lawsuit alleges that Bjorklund “was and still is caused to suffer from severe gastrointestinal issues, as well as other severe and personal injuries which are permanent and lasting in nature, physical pain, and mental anguish.”
In statements, both companies defended their products. Novo Nordisk claimed that gastrointestinal events are “well-known side effects of the GLP-1 class,” according to CBS News. The company added:
“For semaglutide, the majority of GI side effects are mild to moderate in severity and of short duration. GLP-1’s are known to cause a delay in gastric emptying, as noted in the label of each of our GLP-1 RA medications. Symptoms of delayed gastric emptying, nausea and vomiting are listed as side effects.”
And, as reported by The Hill :
“Patient safety is of utmost importance to Novo Nordisk. … We are continuously monitoring the safety profile of our products and collaborate closely with authorities to ensure patient safety, including adequate information on gastrointestinal side effects in the label.”
Eli Lilly said patient safety is its “top priority,” according to CBS News, and the company is “actively engage[d] in monitoring, evaluating and reporting safety information for all our medicines.”
NBC News cited FDA spokesperson Chanapa Tantibanchachai, who said it’s “unclear” whether GLP-1 medications are connected to occurrences of gastroparesis.
A separate FDA statement cited by CNN states the agency has “received reports of gastroparesis with semaglutide and liraglutide, some of which documented the adverse event as not recovered after discontinuation of the respective product at the time of the report.”
‘This medicine made my life hell’
However, lawyers for Bjorklund say that many more patients are ready to come forward with lawsuits of their own.
According to CBS News, Pennock’s firm “is investigating 400 other inquiries from clients across 45 states,” while according to The Hill, Pennock ultimately expects to see “thousands of such cases.”
Several patients also spoke to media outlets regarding their injuries.
Brea Hand told CBS News, “The stomach pain was just unbearable and I couldn’t keep anything down. I would drink something and within minutes, like five, 10 minutes later, I would be throwing up.”
Hand visited the hospital six times while taking Ozempic and was admitted to an intensive care unit. She is not involved in the lawsuit.
A July 25, CNN featured the stories of other patients, including Louisiana resident Joanie Knight, 37, who said “I wish I never touched it. I wish I’d never heard of it in my life,” referring to Ozempic. “This medicine made my life hell. So much hell.”
And Emily Wright, a 38-year-old teacher from Toronto, began taking Ozempic in 2018. Today though, despite not having taken the medication “for a year,” she said, “I’m still not back to my normal.” She told CNN she is now vomiting so frequently that she was obliged to take a leave of absence from her job.
In recent months, several reports have indicated that Ozempic in particular is linked to a range of other health problems.
Last month, health regulators in Iceland, followed by the European Medicines Agency, began investigating reports that Ozempic and other popular weight-loss drugs are linked to the inducement of suicidal thoughts.
Ozempic also was linked to an increased risk of adverse kidney events, diabetic retinopathy, and metabolic, nutritional, eye, retinal, urinary and cardiac disorders.
And in April, the FDA warned that Ozempic should be discontinued at least two months prior to pregnancy because it takes that long for the body to eliminate the drug.
However, those warnings are buried, and long-term testing won’t be completed for years. The drug was not studied in pregnant women during clinical trials.
Michael Nevradakis, Ph.D., based in Athens, Greece, is a senior reporter for The Defender and part of the rotation of hosts for CHD.TV’s “Good Morning CHD.”
Remdesivir may be the most despised drug in American history, earning the nickname Run Death Is Near for its lethal record during COVID. Experts claimed that it would stop COVID; instead, it stopped kidney function, then blasted the liver and other organs. Now this reviled destroyer of kidneys has been approved by the FDA for COVID treatment of kidney patients. Does anybody else feel as if the FDA is shoving its power in our faces and laughing at us?
I’ve been joining online support groups for people who lost loved ones to the Remdesivir Protocol — a nightmarish sequence in which a patient is isolated in the hospital, bullied into taking Remdesivir, ventilated, and then sedated to death. Thousands of Americans were killed this way, possibly hundreds of thousands.
These support groups are a deeply somber business. Grieving faces fill the screen of people who lost a parent, spouse, sibling, or child. Some speak with icy anger; some choke back sobs as they tell of the deadly abuse inflicted on their loved ones, shattering their families forever.
I asked them what they thought of the FDA’s decision to approve Remdesivir for people with severe renal impairment, including dialysis. “Morally, how can you do that?” Joyce Wilson said. “It’s a death sentence. They didn’t care if people had kidney issues or not. My husband went into the hospital in kidney distress. They exacerbated it with Remdesivir. Then they ventilated him, and he died.”
“This is absurd,” Tracy Bird told me. “The FDA can no longer be trusted with any drug under any circumstances. It’s all conflicts of interest. My husband Jeff had strong kidney function when he went in the hospital. They gave him Remdesivir, and three days later, he was in kidney failure.”
“My daughter’s story is no different than anyone else’s,” Denise Fritter said. “Jamie was 36 and looking forward to getting married. The hospital refused to consider any other modalities of treatment for her. They insisted on Remdesivir. Then they put her on a vent and murdered her. I think the FDA is using Remdesivir to fulfill their own agenda.”
Cheri Martin, who lost her husband Steven to the protocol, chimed in with thoughts on the agenda: “They’re going to use this decision as a way to clean house of renal patients and people on dialysis. It’s saving a ton of money for Medicare over the next twenty years.”
“I can’t believe the FDA would approve this,” MaryLou said. “My son was 37 years old. He went into the hospital with two blood clots, but his kidneys were functioning. They gave him Remdesivir, and in twelve hours, his kidneys stopped working, and his organs began to fail. We never saw him open his eyes again.”
Michelle Conway said, “I took my husband to the E.R., and the next day, they told me he was going on Remdesivir. I said absolutely not. I wanted him on other treatments, but they refused all of it. They isolated him and told him he had to have Remdesivir or he’d die, and he agreed. I got to watch his last rites over a video conference. I know he was murdered by Remdesivir.”
A woman I’ll call Maya joined the support group for the first time to share her story. She’s a survivor of the hospital protocol, and there aren’t many of those. “I refused Remdesivir, and I refused the ventilator. But they find other ways to take you out. The doctors were pissed at me. They called my husband to pressure him. They fear-monger you with all these lies. And they pull your loved ones away from you. I was all by myself trying to make decisions.”
The discussion often turned to the weird carelessness and indifference to standard medical procedures in the hospitals during COVID. “Multiple times in my husband’s record, it said he was not a candidate for Remdesivir,” Lisa said. “They gave it to him anyway, and he went into renal failure and died.”
“The Remdesivir fact sheet clearly states that it may cause kidney and liver failure. And that’s exactly what happened to my husband Richard,” Michelle Strassburg said. “They’re doubling down on this preposterous decision. I’m at a loss for words.”
“It’s so important that in their own literature of Remdesivir, they state that it’s supposed to be given early,” Catherine said. “Yet they kept stalling my husband. They sent him home and said to sign up for monoclonal antibodies. But when he showed up for it, they said they were too backed up. By the time he was hospitalized, he was really sick. They gave him Remdesivir, and he had a stroke.”
Everyone in the group knows about the financial incentives that drove the hospital’s insistence on Remdesivir. The federal government paid hospitals a staggering 20% bonus on the entire hospital bill of patients treated with Remdesivir. They also handed out lavish extra payments for ventilating patients. And, perhaps most tellingly, the feds rewarded hospitals with more money for patients who died of COVID instead of those who were healed.
Gregory Gandrud, the treasurer of the California Republican Party, understands financial incentives well. He explained the money behind his hospitalization. “They gave me $37,000’s worth of Remdesivir, but it obviously didn’t help because I wound up on a ventilator. My hospital bill was $920,000 for the 44 days I was there. Nobody offered me ivermectin, which is cheap, effective, has no side effects, and you can take at home.”
Many in the group expressed frustration at trying to get justice. The PREP Act indemnified medical institutions from any actions they took during the federally declared COVID emergency. Lawyers are reluctant to take cases because they don’t see how to break through the hospitals’ indemnity shield.
After the support group, I spoke with Jamie Scher, who told me that her legal team was ready to file a complaint against Gilead today. Gilead is the lucky maker of Remdesivir, enjoying fabulous profits from this previous loser of a drug, which turned into a billion-dollar winner during COVID.
Jamie said she has over 1,000 plaintiffs, and, unfortunately, the list is growing daily. She’s working hard to raise funds for the lawsuit; people interested in finding out more can visit her website at myerandscher.com.
Another way to circumvent the PREP Act may be to get malpractice insurance carriers to not insure hospitals and doctors for the use of this protocol and lethal drugs like Remdesivir. Jamie said prosecutors could then hold them accountable for intentionally killing people, knowing that these drugs do not help; they only harm.
I confess that after these support groups, I find it difficult to sleep. I keep reliving the anguish of these wonderful people. “They think we’re stupid,” I hear Erin say. Denise’s sobs echo in my head, as she cries, “Why did God take my daughter from me? I’ll never know.” But her voice strengthens as she adds, “I do know we’re all warriors in a spiritual battle.” And Catherine offers words of hope: “Despite it all, I believe we’re going to get justice.”
In a move that defies all regulatory convention, the U.S. Food and Drug Administration (FDA) approved a supplemental new drug application (sNDA) for the use of Veklury® (remdesivir) in COVID-19 patients with severe renal impairment, including those on dialysis. With this approval, Veklury is now the first and only approved antiviral COVID-19 treatment that can be used across all stages of renal disease but has no efficacy data to support its administration.
The phase 3 REDPINE trial failed to recruit sufficient subjects to assess efficacy. Instead of properly rejecting the application, the FDA went ahead and approved the drug with insufficient safety and efficacy data. The drug has struggled in recent years as patients commonly decline the antiviral since the November, 2020, WHO warning against inpatient use. Remdesivir can cause both kidney injury and liver damage, thus with no mortality benefit, many believe it should not be used.
The FDA approval action defies logic and will be added to a long list of acts that will be considered malfeasance and will be up for review when the commissioner and agency is finally called to justice.
A 13,685-page tranche of documents related to Moderna’s COVID-19 vaccine clinical trials released Tuesday contain details about the deaths of 16 trial participants, the prevalence of severe adverse events (SAEs) and other abnormalities.
The documents, previously submitted by Moderna to the U.S. Food and Drug Administration (FDA) as part of the licensing process for Moderna’s Spikevax COVID-19 vaccine, also exposed an “utter lack of thoroughness” in how the trials were conducted, according to Defending the Republic (DTR), a Dallas-based nonprofit that obtained the documents via a a still-pending Freedom of Information Act lawsuit against Moderna.
The documents, shared with The Defender in advance of their public release, are the first set of “Moderna documents” to be released as part of the lawsuit — with approximately 8,000 more pages expected to follow later this year.
Travis Miller, a Fort Worth-based attorney representing DTR, told The Defender, “These documents include over 13,500 pages relating to serious adverse event listings that document injuries — such as shingles and Bell’s palsy and other more serious conditions — which we believe may be related to the Moderna COVID-19 vaccine.”
DTR also received documents describing experiments involving mRNA injections on rats in 2017-2018, prior to the onset of COVID-19. Miller told The Defender these studies revealed fetal abnormalities in pregnant rats.
Dr. Meryl Nass, an internist, biological warfare epidemiologist and member of the Children’s Health Defense scientific advisory committee, said the Moderna clinical trial data bear similarities to the outcomes seen in the Pfizer COVID-19 vaccine trials, and raise several questions about safety and liability.
Nass told The Defender :
“Both the preclinical (animal) studies of Moderna and of Pfizer revealed skeletal abnormalities in the offspring of vaccinated mice and rats at higher-than-normal rates and revealed vaccine components travelled throughout the body into all organs.
“Both the Pfizer and Moderna trial data in humans reveal concerning deaths and side effects that were attributed to other causes, but likely were vaccine side effects.”
Nass said “it appears” the FDA did not perform due diligence regarding the clinical trials for each vaccine.
“Did the FDA perform its required regulatory function to oversee the conduct of the trials?” Nass asked. “Or did Operation Warp Speed wave the vaccines through without a proper FDA review of the data?”
Nass asked “who is responsible” if the FDA failed to “perform its regulatory tasks?”
She said the clinical trial data also lead to questions about the liability shield enjoyed by vaccine manufacturers:
“If Moderna (and Pfizer) knew of more problems with the vaccines than they acknowledged, will they have liability under the PREP Act?
“Finally, pilot lots of vaccine (used for the clinical trials of the Pfizer vaccine, and likely the Moderna vaccine) were considerably different than lots made later, using different methods. This was noted by the European Medicines Agency.
“Therefore, do the clinical trial findings even apply to everyone else who received the vaccine later?”
Serious adverse events routinely classified as ‘unrelated’ to vaccine
Miller criticized Moderna’s lack of scientific rigor in determining the causes of the deaths and adverse events, saying that, in several cases, “Individuals who died after receiving the Moderna vaccine were not given an autopsy.”
According to DTR, “The study’s authors indicated that of those 16 deaths, only two autopsies were performed, five of the dead were not autopsied, and the autopsy status of nine of the dead was ‘unknown.’”
In one instance, a 56-year-old woman experienced “sudden death” 182 days after receiving her second dose of the Moderna vaccine. The cause of death was listed as “unknown” and no autopsy was performed.
“It seems they purposely decided not to investigate suspicious deaths in case the Moderna vaccine might be the cause,” DTR stated in its summary.
Yet the deaths “did not stop those running these ‘studies’ from concluding, despite the absence of evidence, that the Moderna vaccine was not related to these deaths,” DTR added.
Several trial participants also developed neurological disorders, DTR said. “One 44-year-old female had ‘left side facial paralysis’ just eight days after the second dose” and “Numerous vaccinated participants saw the onset of shingles less than 10 days after vaccination.”
This was not the full extent of SAEs sustained by trial participants. According to DTR:
“Subsequent analyses of reports from the FDA VAERS [Vaccine Adverse Events Reporting System] database, the Department of Defense’s DMED [Defense Medical Epidemiological Database], and European regulators showed heightened rates of these illnesses following administration of the Moderna vaccine.”
Similar to the Pfizer documents released last year, the Moderna documents indicate SAEs were routinely classified as being “unrelated” to the vaccine. According to DTR:
“… similar to their treatment of deaths post-vaccination, the studies seemed predestined to conclude that these serious adverse events — many of them life-threatening — were not related to the Moderna vaccine. It didn’t matter whether the adverse event occurred within days of vaccination.
“All this creates serious doubt about the safety of the Moderna vaccine and the standards by which it was approved by the FDA,” Miller said.
According to DTR, the documents also contained “troubling” evidence from animal studies.
Referring to the results of one study, DTR stated, “The findings of this study are troubling: the mRNA vaccine altered the skeletal variations of the rat fetuses and the ‘female pregnancy index’ of the vaccinated rats was significantly lower than the control group.”
Other abnormalities noted in this study included an above-average rate of “common skeletal variations consisting of wavy ribs and increase[d] nodules,” a “statistically significant higher” mean number of reproductive cycle lengths and a lower incidence of mating and pregnancy in the mRNA-1273 group rats compared to the control group.
Moderna included an older study, conducted in 2017 and 2018, prior to the COVID-19 pandemic, in its application for FDA approval. The study showed similar results, with mRNA found in several organs.
According to DTR, “Testing revealed that ‘mRNA-1647 was detected in all of the analyzed tissues except for kidney[s],’ with elevated levels of mRNA-1647 found in the spleen and eye. Notably, mRNA-1647 was detected in the brain and heart.”
FDA twice denied FOIA requests for release of the Moderna documents
Miller told The Defender that DTR sued Moderna after the FDA “wrongly denied our request for the expedited production of the records submitted by Moderna in support of its Biologics License Application (BLA) for its COVID-19 vaccine ‘Spikevax.’”
DTR said it reached an agreement earlier this year with the FDA for the production of approximately 24,000 pages of some of the most important records submitted by Moderna in support of its Biologics License Application.
The agreement, announced March 31, required the FDA to release the first 13,685-page set of documents by July 17, and the remainder by the end of 2023.
However, the FDA twice rejected DTR’s request — first on Feb. 9, 2022 and again on June 6, 2022 — claiming DTR had not shown “a compelling need for expedited processing” of the documents. DTR sued the FDA the following day.
While the lawsuit is still ongoing, Miller told The Defender it will be “dismissed per agreement by the parties” once the FDA provides the remaining documents.
According to Miller, these documents include:
Moderna’s May 28, 2021 original application.
Postmarketing reports of herpes zoster.
Data related to unsolicited adverse events.
Data relating to analysis and efficacy against severe COVID.
Information on antibody quantification.
Information on postmarketing vaccine effectiveness.
The documents are expected to be released by the end of the year.
Miller told The Defender he hopes the findings in the Moderna documents will “at a minimum, lead to further Congressional oversight of the FDA’s approval process and for accountability within that agency.”
Michael Nevradakis, Ph.D., based in Athens, Greece, is a senior reporter for The Defender and part of the rotation of hosts for CHD.TV’s “Good Morning CHD.”
Introduction: For the greater part of a decade the US, the UK and the EU have been carrying out a campaign to undermine and overthrow the Russia government and in particular to oust President Putin. Fundamental issues are at stake including the real possibility of a nuclear war.
The most recent western propaganda campaign and one of the most virulent is the charge launched by the UK regime of Prime Minister Theresa May. The Brits have claimed that Russian secret agents conspired to poison a former Russian double-agent and his daughter in England, threatening the sovereignty and safety of the British people. No evidence has ever been presented. Instead the UK expelled Russian diplomats and demands harsher sanctions, to increase tensions. The UK and its US and EU patrons are moving toward a break in relations and a military build-up.
A number of fundamental questions arise regarding the origins and growing intensity of this anti-Russian animus.
Why do the Western regimes now feel Russia is a greater threat then in the past? Do they believe Russia is more vulnerable to Western threats or attacks? Why do the Western military leaders seek to undermine Russia’s defenses? Do the US economic elites believe it is possible to provoke an economic crisis and the demise of President Putin’s government? What is the strategic goal of Western policymakers? Why has the UK regime taken the lead in the anti-Russian crusade via the fake toxin accusations at this time?
This paper is directed at providing key elements to address these questions. … continue
This site is provided as a research and reference tool. Although we make every reasonable effort to ensure that the information and data provided at this site are useful, accurate, and current, we cannot guarantee that the information and data provided here will be error-free. By using this site, you assume all responsibility for and risk arising from your use of and reliance upon the contents of this site.
This site and the information available through it do not, and are not intended to constitute legal advice. Should you require legal advice, you should consult your own attorney.
Nothing within this site or linked to by this site constitutes investment advice or medical advice.
Materials accessible from or added to this site by third parties, such as comments posted, are strictly the responsibility of the third party who added such materials or made them accessible and we neither endorse nor undertake to control, monitor, edit or assume responsibility for any such third-party material.
The posting of stories, commentaries, reports, documents and links (embedded or otherwise) on this site does not in any way, shape or form, implied or otherwise, necessarily express or suggest endorsement or support of any of such posted material or parts therein.
The word “alleged” is deemed to occur before the word “fraud.” Since the rule of law still applies. To peasants, at least.
Fair Use
This site contains copyrighted material the use of which has not always been specifically authorized by the copyright owner. We are making such material available in our efforts to advance understanding of environmental, political, human rights, economic, democracy, scientific, and social justice issues, etc. We believe this constitutes a ‘fair use’ of any such copyrighted material as provided for in section 107 of the US Copyright Law. In accordance with Title 17 U.S.C. Section 107, the material on this site is distributed without profit to those who have expressed a prior interest in receiving the included information for research and educational purposes. For more info go to: http://www.law.cornell.edu/uscode/17/107.shtml. If you wish to use copyrighted material from this site for purposes of your own that go beyond ‘fair use’, you must obtain permission from the copyright owner.
DMCA Contact
This is information for anyone that wishes to challenge our “fair use” of copyrighted material.
If you are a legal copyright holder or a designated agent for such and you believe that content residing on or accessible through our website infringes a copyright and falls outside the boundaries of “Fair Use”, please send a notice of infringement by contacting atheonews@gmail.com.
We will respond and take necessary action immediately.
If notice is given of an alleged copyright violation we will act expeditiously to remove or disable access to the material(s) in question.
All 3rd party material posted on this website is copyright the respective owners / authors. Aletho News makes no claim of copyright on such material.