HHS to ‘Revolutionize’ Vaccine Injury Compensation, RFK Jr. Tells Tucker Carlson
By Suzanne Burdick, Ph.D. | The Defender | July 1, 2025
Health Secretary Robert F. Kennedy Jr. sat down yesterday with Tucker Carlson to share an update on his mission to end the skyrocketing rate of autism in U.S. kids.
By the end of their nearly 90-minute conversation, the two had covered a slew of topics, including pharmaceutical ads on TV, increasing compensation for the vaccine-injured, and the need for a “truth commission” to uncover who and what caused the COVID-19 pandemic.
Carlson, who last year left FOX News after being the network’s “most popular host,” now runs “The Tucker Carlson Show.” He broke his interview with Kennedy into five “chapters”:
- Uncovering the Reason for Skyrocketing Rates of Autism
- Is It Possible to End the Corrupt Relationship Between Big Pharma and Corporate Media?
- Will There Be Compensation for the Vaccine-Injured?
- RFK’s Firing of So-Called “Experts”
- The Real Reason Fauci Got a Pardon
Below are highlights from each.
HHS will do honest, open research on autism and vaccines
In the past, the Centers for Disease Control and Prevention (CDC) failed to honestly and adequately research the possible link between vaccines and autism, Kennedy said.
The CDC ignored recommendations from the Institute of Medicine to do a “litany” of studies to get at the issue, Kennedy said, including animal models, observational studies, bench studies and epidemiological studies.
“But what we’re going to do now,” he said, “is we’re going to do all the kinds of studies that the Institute of Medicine originally recommended.”
The National Institutes of Health (NIH) in April announced a new research program to study what causes autism and why autism diagnoses are on the rise.
NIH will make data from Medicare and Medicaid available to independent scientists for analysis. Data from the Vaccine Safety Datalink — a huge repository for health records — will also be used, Kennedy said.
Raw data will be made available to the public whenever possible, Kennedy said.
“Something new that we’re bringing in is that every study will be replicated,” he added.
Big Pharma ads fail to benefit patients and doctors
Sens. Bernie Sanders (I-Vt.) and Angus King (I-Maine) last month introduced federal legislation to end direct-to-consumer prescription drug advertising.
Kennedy didn’t reference the bill or say he supported a ban on such ads. However, he outlined several reasons why pharmaceutical marketing on mainstream media is bad for public health.
Many ads are misleading, he told Carlson. “Even the music and the video, the photos that they show … it’s sending a message that if you take this drug, you’re going to be riding jet skis and playing volleyball and water skiing and have a great-looking spouse.”
Meanwhile, the ads feature the most expensive version of the drug rather than the generic version.
“They’re not going to advertise the generics because they’re not making any money,” Kennedy said. “So they’re advertising the ones that are the highest profit margins for them.”
Plus, the U.S. taxpayer bears the brunt of the cost while the drug company profits. Kennedy explained:
“Normally, if you see an advertisement on TV like for Coca-Cola, you then have a choice to go get that and you’re paying out of your pocket for it.
“When somebody buys a pharmaceutical drug, it’s Medicaid and Medicare that are paying for it … it’s the taxpayer. … And we’re paying for the ads because they’re tax-deductible.”
When a patient sees the ad and asks a doctor for the drug, the doctor — who is told by a “corporate bean counter” to limit time with a patient to only 11 minutes — has to choose whether to use the time trying to talk the patient out of the drug, Kennedy said. But if the doctor does that, the patient likely goes away unsatisfied.
Or the doctor could just say, “All right, you want this prescription? I’ll write it for you.” Then the patient will be satisfied and come back, Kennedy said. “The doctors hate it. … And nobody thinks that this is good for public health. It is hurting us.”
Kennedy said the censorship of vaccine-related information on social media is also a problem.
The U.S. Supreme Court yesterday denied Children’s Health Defense’s (CHD) petition to hear its censorship case against Meta, the parent company of Facebook.
CHD sued Meta in August 2020 and filed an amended complaint in November 2020, alleging that government actors partnered with Facebook to censor CHD’s speech — particularly speech related to vaccines and COVID-19 — that should have been protected under the First Amendment. The company deplatformed CHD from Facebook and Instagram in August 2022 and has not reinstated the accounts.
Censorship of scientific results that are critical of vaccines is also a problem, Kennedy added.
Kennedy’s plans to expand vaccine injury compensation program
The National Childhood Vaccine Injury Act of 1986, which granted legal immunity to vaccine makers and created the National Vaccine Injury Compensation Program, also made it difficult for anyone injured by a vaccine to obtain compensation.
“We just brought a guy in this week who is going to be revolutionizing the [National] Vaccine Injury Compensation program,” Kennedy said.
“We’re looking at ways to enlarge the program so that COVID vaccine-injured people can be compensated … we’re looking at ways to enlarge the statute of limitations,” Kennedy told Carlson.
It’s currently limited to three years. “A lot of people don’t discover their injuries till after that,” Kennedy said.
The program has other flaws, including that it has no discovery process, no rules of evidence and historically had corrupt leadership.
“We’re going to change all that,” Kennedy said. “I’ve brought in a team this week that is starting to work on that.”
Kennedy also said HHS will use AI (artificial intelligence) to track vaccine injuries more effectively. The agency plans to use AI in other ways, too, such as speeding up drug approval processes and detecting fraud.
Why CDC vaccine advisory committee needed a clean sweep
Kennedy defended his recent move to fire all members of the CDC’s vaccine advisory panel, saying the board had become “a sock puppet for the industry that it was supposed to regulate.”
On June 11, Kennedy named eight researchers and physicians to the Advisory Committee on Immunization Practices (ACIP), two days after removing all 17 of the previous ACIP members.
“This was a long time coming, Tucker,” Kennedy said. He gave an example to illustrate the kind of financial conflict of interest that had plagued the board for years.
Years ago, the committee approved adding a rotavirus vaccine to the childhood immunization schedule, he said.
Four of the five committee members had “direct financial interest in the rotavirus vaccine,” Kennedy said. “They were working for the companies that made the vaccine, or they were receiving grants to do clinical trials on that vaccine.”
Within a year, that specific rotavirus vaccine was linked to “disastrous” disease in kids and pulled from the market. It was replaced by a different rotavirus vaccine that then-committee member Dr. Paul Offit had helped develop.
“Then [Offit] and his business partners, Dr. Stanley Plotkin, and a couple of other people, sold that vaccine to Merck for $186 million,” Kennedy recalled.
According to Kennedy, Offit told Newsweek that he won the lottery. “It’s been said of him that he voted himself rich, so that kind of conflict was typical on that committee.”
Could a ‘truth commission’ hold Fauci accountable?
Carlson and Kennedy discussed the origins of COVID-19 and the possible reasons for Dr. Anthony Fauci’s presidential pardon.
Just before leaving office, former President Joe Biden preemptively pardoned Fauci. The pardon, retroactive to Jan. 1, 2014, addresses “any offenses” Fauci committed during this period, including in his former capacities as director of the National Institute of Allergy and Infectious Diseases, member of the White House COVID-19 Response Team and chief medical adviser to Biden.
When Carlson pressed Kennedy to comment on Fauci’s motivations for funding coronavirus research in China, Kennedy said he tried to avoid speculation.
That’s why in his book, “The Real Anthony Fauci,” he reports only what Fauci did, not Fauci’s possible motivations, he said.
Carlson said, “It sounds like Fauci is beyond the reach of the law at this point.”
Kennedy responded, “Yeah, I think generally, unless there was a truth commission, you know, which they did in South Africa. They did it in Central America after the 1980s wars there, and they were very, very helpful to those societies. I think we should probably do something like that now.”
Kennedy explained how a truth commission works:
“You have a commission that hears testimony on what exactly happened. Anybody who comes and volunteers to testify truthfully is then given immunity from prosecution. But so that at least the public knows who did what. …
“People who are called and don’t take that deal and perjure themselves, they then can be prosecuted criminally.”
This article was originally published by The Defender — Children’s Health Defense’s News & Views Website under Creative Commons license CC BY-NC-ND 4.0. Please consider subscribing to The Defender or donating to Children’s Health Defense.
“Why Can’t We Talk About This?”
Rainey Media TV | June 4, 2025
Please Support Our Film via: E-Transfer: dean_rainey@yahoo.ca
PAYPAL: deanrainey@raineymedia.com
Buy My A Coffee: https://buymeacoffee.com/raineymedia
SNAIL MAIL: Rainey Media, PO Box 5, Delhi PO Main, ON, N4B 2W8 Canada
To book your own screening: deanrainey@raineymedia.com
“Why Can’t We Talk About This?” delves into the life of a man grappling with the aftermath of a COVID-19 vaccine injury, weaving his personal struggle into a broader examination of why such experiences are rarely discussed.
To help Michael via support: https://www.gofundme.com/f/benefit-fo…
We’ve made this film easily accessible for everyone because the information it contains and the discussion it starts is just too important. This film had no funding and was made without sponsorship. All costs were paid out of my own pocket. My team and I spent over a year and a half making this film. Any support you give will go towards expenses incurred making and marketing the movie. (Even sending the price of a movie ticket would help.) Once we cover those costs, we will be providing Michael with a share of the proceeds.
To order DVDs of this film, visit: https://raineymedia.com/video-store/
Also available on Rumble.
‘Every American Wearing a Wearable’ Is Not a Vision We Share
By Children’s Health Defense EMR & Wireless Team | The Defender | June 26, 2025
During recent congressional testimony, U.S. Health and Human Services Secretary Robert F. Kennedy Jr. stated:
“We’re about to launch one of the biggest advertising campaigns in HHS history to encourage Americans to use wearables … my vision is that every American is wearing a wearable within four years.”
Kennedy was responding to a question about whether consumers should continue to have access to wearables. He explained that wearables allow users to constantly track in real-time how food and lifestyle choices affect their health metrics. He also claimed that wearables are key to the Make America Healthy Again (MAHA) agenda.
We agree that people should be able to monitor their health in innovative ways using the technology they choose. But we do not think the federal government should try to push wearables on every American.
A wearable is an electronic device — such as a smartwatch, fitness tracker or smart ring — worn on the body. It’s made up of dozens of sensors and wireless technologies that continuously collect, monitor and transmit biometric and other sensitive data.
“We do not share this vision,” said Miriam Eckenfels, director of the Children’s Health Defense (CHD) Electromagnetic Radiation (EMR) & Wireless Program. “Quite the contrary, we oppose governmental pressure to incentivize the widespread use of wearables. They pose serious health risks, especially to children, and they threaten privacy.”
Wireless technologies, including wearables, have clear and well-documented harms. These devices continuously emit radiofrequency (RF) radiation in direct contact with the body for long periods of time.
They also have multiple transmitters/receivers (Bluetooth, Wi-Fi and cellular), operating on several different radio bands. Cumulative and long-term exposures have known significant risks.
RF radiation exposure is associated with a wide range of adverse health effects, including “increased cancer risk, cellular stress, increase in harmful free radicals, genetic damages, structural and functional changes of the reproductive system, learning and memory deficits, neurological disorders, and negative impacts on general well-being.”
A systematic review commissioned by the World Health Organization (WHO) last month concluded that there is “high certainty” evidence that cellphone radiation exposure causes two types of cancer in animals.
Higher-frequency millimeter wave (MMW) transmissions used in 5G cellular networks are also known to produce eye damage and skin burns. An industry study by the Institute of Electrical and Electronics Engineers (IEEE) concluded that overexposure to MMW is expected to produce burns “like those produced when a person touches hot objects or flames.”
Children, pregnant women at even greater risk
Children have smaller bodies, developing nervous systems, more conductive tissue and longer lifetimes of exposure compared to adults, putting them at even greater risk of harm from radiation exposure. Their cells are dividing and growing at a higher rate, so DNA damage is magnified.
Other vulnerable populations include pregnant women, and people with implanted devices, chronic health conditions and Electromagnetic Radiation Syndrome (EMR-S).
The U.S. Food and Drug Administration (FDA) has issued official guidance cautioning that individuals with pacemakers and other implanted medical devices should keep wearables like smartwatches at a distance due to potential interference and malfunction. Manufacturers like Apple also include guidelines and warnings for wearables.
This highlights a broader point: wearables are not safe or suitable for “every American.”
In 2021, CHD won a landmark case against the Federal Communications Commission (FCC), in which the U.S. Court of Appeals for the District of Columbia Circuit ruled that the FCC had failed to consider extensive evidence of harm from wireless radiation.
The court found that the FCC did not consider peer-reviewed scientific research on the harmful effects of wireless radiation exposure on children, the brain and nervous system, male fertility and people with EMR-S.
The ruling specifically cited the agency’s failure to address studies showing oxidative stress, DNA damage, and the health risks from modulation and cumulative exposure.
The court also ordered the agency to explain how its limits are protective. Yet almost four years later, the FCC has still not complied.
This ruling validated years of scientific evidence about the harms of wireless technology and confirmed that the public, including wearable users, continues to be actively misguided by industry and government agencies alike.
Biometric data collection raises privacy concerns
Wireless technologies also have extensive and well-documented privacy impacts. They continuously collect biometric data, including heart rate, quality of sleep, blood pressure, cholesterol levels, oxygen levels, calorie burn, sweat gland emissions, hormone levels, body temperature, emotional responses, movement and precise geolocation.
This biometric data is transmitted over the internet and can be used to create an intimate profile of the user’s physical and psychological states. This intimate profile can be made available to employers, medical providers, private corporations, artificial intelligence systems, insurance companies and government entities.
This surveillance infrastructure may lay the groundwork for psychological targeting, predictive modeling, social control and unprecedented intrusions into personal freedom.
These risks cannot be left out of any discussion of so-called “digital health.”
“We are eager to learn more about Secretary Kennedy’s full intent regarding wearables,” said Eckenfels. “The growing push for widespread adoption of wearables, which exposes users to constant RF radiation in direct contact with the body, is concerning and fundamentally at odds with the values of informed consent, privacy and bodily autonomy that CHD defends.”
Eckenfels added:
“The public deserves radical transparency about wearables’ health and privacy risks. Their use must remain a personal choice and not a public health objective. We do not share — indeed, we oppose — a vision where everyone is subject to constant wireless exposure in direct contact with the body and biometric tracking.
“What amounts to technocratic surveillance should not be normalized, encouraged and promoted at the federal level.”
This article was originally published by The Defender — Children’s Health Defense’s News & Views Website under Creative Commons license CC BY-NC-ND 4.0. Please consider subscribing to The Defender or donating to Children’s Health Defense.
Vaccine Makers Signal Fear Over Removal of Neurotoxic Injected Aluminum Ingredient
By Jefferey Jaxen | June 27, 2025
Former FDA commissioner Dr. Scott Gottlieb made his ceremonial appearance on CNBC to titrate the public well with Big Pharma talking points in the wake of this week’s ACIP meeting.
After speaking for less than one minute and forty seconds, Gottlieb used the tired, inaccurate slur ‘anti-vaxxer’ four times in a failed attempt to frame an us verse them narrative like it was 2015 again.
With the newly appointed ACIP committee vote to remove the mercury-based preservative thimerosal from the few remaining flu shots, Gottlieb wasted no time circling the wagons to protect the widespread, problematic aluminum adjuvant in several vaccines on the CDC’s childhood schedule.
His concern was that would be ACIP’s next target. And he’s probably right.
“This is a very safe ingredient” stated Gottlieb regarding the regular injection of aluminum nanoparticles into infants, children and adolescents at scale.
Zero pushback or questions from the interviewer to challenge him per usual.
How settled is the safety science of injecting aluminum in children?
The Informed Consent Action Network sent a legal letter to the U.S. Centers for Disease Control and Prevention (CDC) and the National Institutes of Health (NIH) in 2019 demanding any human or animal studies relied upon by these agencies to establish the safety of injected aluminum.
The agencies produced no documents nor could they located a single study showing the safety of aluminum in childhood vaccines.
Meanwhile, a 2021 study published in the Journal of Trace Elements in Medicine and Biology found that six childhood vaccines contain a statistically significant greater quantity of aluminum adjuvant than is provided for on these products’ labeling. This study promped ICAN to demand the FDA assure that vaccine manufacturers are disclosing accurate information regarding the amount of aluminum adjuvant in their childhood vaccines. The agency has since stonewalled the request.
Here’s the embarrassing, anti-scientific part Gottlieb forgets to mention.
The rationale for injecting aluminum adjuvant nanoparticles into newborns was allowed and justified by a single 2011 study, by a single FDA scientist named Dr. Robert Mitkus.
Author J.B. Handley in his book How to End the Autism Epidemic writes the following about Mitkus’ study:
What would be lost on the average layperson is that the only biological science Dr. Mitkus considered in making his safety assessment was a single study that infused (rather than injected) aluminum citrate (rather than aluminum hydroxide) into adults (rather than babies). It’s hard to put this seemingly minor detail in proper context. In no other drug on the planet (except for vaccines) would safety standards ever be determined without using the actual product (aluminum hydroxide) administered in the proper way (intramuscular injection), into the proper patient population (infants).
World-renowned researchers called out this fact in their 2018 study by stating:
“To date, aluminum adjuvants per se have, perhaps surprisingly, not been the subject of any official experimental investigation, and this being in spite of the well-established neurotoxicity of aluminum.”
Will aluminum adjuvants be ACIP’s next target? Are studies being commissioned by Jay Bhattacharya’s NIH to look at these ingredients and their well-established role in creating chronic disease in American Children? All open questions at the time of this writing.
As for Scott and his industry pals, shots across the bow appear to be signaling that it’s no longer business as usual.
Gottlieb left his position as FDA commissioner only to accept a position on the board of Pfizer in less than three months.
Gottlieb is Big Pharma’s jack-in-the-box who seems to pop up and make noise at key moments when public pressure is applied which threatens bottom line profit margins of their liability-free injectable product lines.
His corporate media residency at CNBC allows for rapid response industry talkings points to roll from his mouth at a moment’s notice whenever his handlers decide to make him dance.
Prior to the pandemic, as questions swirled about a connection between vaccines and autism, Gottleib was there. When asked by the CNBC reporter why parents claim that their children developed autism or “something on the spectrum” right around the time they received their shots, Gottlieb blamed coincidence by saying:
“Children who are gonna display symptoms of autism and other developmental disorders, those start to manifest and become self-evident right around the time kids are getting vaccinated.”
Magic. Like Fauci, Gottleib is his own version of The Science™. What he says is ordained, never questioned during interviews. A continuous appeal to authority. Why? Because Pfizer man said so.
With a little luck, revolving door riders like Gottlieb will be artifacts of a shameful past era. Where U.S. regulatory agencies continually launched their leaders into the waiting arms of the industries they regulated.
‘Between a Shot and a Hard Place’: Autism, Vaccines and the Illusion of Certainty
By Dr. Joel ‘Gator’ Warsh | The Defender | June 25, 2025
For years, the public has been told the vaccine-autism question is closed — case dismissed, myth debunked, science settled.
But when you peel back the headlines and actually examine the evidence, a startling truth emerges: We haven’t really studied the question at all. Not thoroughly. Not independently. Not with the urgency or integrity the issue demands.
The most commonly cited research? A handful of studies on the MMR vaccine and thimerosal, a mercury-based preservative that was largely removed from childhood vaccines over two decades ago. That’s it.
No comprehensive analysis of the full vaccine schedule. No robust long-term comparisons between vaccinated and unvaccinated children. No meaningful investigation into the timing, combinations, or cumulative biological impact of dozens of shots now given in infancy and early childhood.
In other words, we haven’t looked. And yet we claim to know.
As a pediatrician with formal training in epidemiology, I approached the research with trust in the system and confidence in the data. But what I encountered while investigating for my book, “Between a Shot and a Hard Place,” left me stunned.
I expected to uncover a vast body of high-quality science — long-term trials, robust safety evaluations, rigorous comparisons between vaccinated and unvaccinated children.
Instead, I found a shallow pool of studies — many small, some outdated, most narrowly focused on just one vaccine. There was no comprehensive scrutiny of the full schedule, no real curiosity about timing, interactions, or vulnerable populations.
It wasn’t that the science had disproven a link — it’s that the science had barely asked the question. And that silence speaks volumes.
We cannot claim certainty where inquiry has been suppressed. We cannot dismiss parent experiences as coincidences when they follow the same patterns again and again.
And we cannot afford to confuse lack of evidence with evidence of safety. The stakes are too high — and our children deserve better.
The rise in autism, and the refusal to ask why
Autism now affects 1 in 31 children in the U.S., with rates as high as 1 in 12.5 boys in California. The increase in diagnoses isn’t just about better awareness — more children today are deeply affected, with significant developmental and intellectual disabilities.
This is a public health crisis. Yet somehow, asking whether vaccines might play a role is taboo.
Parents see the change firsthand. A baby babbles, smiles, and makes eye contact — then suddenly, after a routine doctor visit, that progress stops. Words disappear. Eye contact fades. Regression sets in.
These stories follow a pattern, and while correlation is not causation, patterns are where science begins. But instead of investigation, we dismiss these parents. Instead of listening, we silence them.
The research we’re missing
I combed through decades of vaccine safety literature. The results were sobering.
- There are no long-term, large-scale studies comparing fully vaccinated children to unvaccinated ones using standardized developmental assessments.
- No comprehensive evaluation exists of the full CDC vaccine schedule as administered in real life.
- Most studies focus narrowly on the MMR vaccine or thimerosal, a mercury-based preservative largely removed from pediatric vaccines two decades ago.
Even the Institute of Medicine acknowledged in a 2013 report that the safety of the full childhood vaccine schedule — especially its timing, spacing, and cumulative exposure— had not been rigorously studied.
If vaccines were a pharmaceutical drug administered in 70 doses before kindergarten, with a suspected link to any chronic disease, we’d demand independent oversight, transparent trials, and long-term tracking.
But because these are vaccines, we declare the science “settled” without proving that it is.
Buried data, ignored whistleblowers
In my research, I came across the 2010 study by Gallagher and Goodman that found a higher autism risk in boys who received the hepatitis B vaccine at birth. It wasn’t widely publicized or followed up.
More disturbing was the 2014 revelation by William Thompson, Ph.D., a senior scientist at the Centers for Disease Control and Prevention who admitted that his team omitted key data in a pivotal MMR-autism study — data that showed increased risk in African American boys. The study was never corrected.
How can we claim the science is settled if major findings are buried and whistleblowers ignored?
A path forward
The vaccine-autism debate won’t be resolved by censorship or soundbites. It will be resolved by doing the science we’ve avoided for too long.
If we truly care about children’s health — and public trust — then we must stop circling the same studies and start asking better questions. That means:
- Funding large, independent, open-label prospective studies comparing fully vaccinated, partially vaccinated, and unvaccinated children — evaluating real-world vaccine schedules, not just single shots in isolation.
- Studying combinations, timing, and aluminum adjuvants using updated toxicology, neurodevelopmental, and immunological tools.
- Taking parental reports seriously as part of observational data—treating them not as “anecdotes to dismiss” but as signals to investigate.
- Removing all financial conflicts of interest from vaccine safety research and creating full transparency for both data and funding sources.
This isn’t about choosing sides. It’s about restoring balance. We can demand rigorous, independent science without being “anti-vax.” We can protect children and respect parental intuition.
But we can’t do either if we keep denying the blind spots in our current system.
To move forward, we must be honest about what we know — and courageous enough to admit what we don’t. Because when it comes to our children’s long-term neurological health, vague reassurances are not enough.
No, the science is not settled. And it’s time we stopped saying it is.
Dr. Joel “Gator” Warsh is a board-certified pediatrician, specializing in integrative and holistic medicine, and the author of “Between a Shot and a Hard Place.”
This article was originally published by The Defender — Children’s Health Defense’s News & Views Website under Creative Commons license CC BY-NC-ND 4.0. Please consider subscribing to The Defender or donating to Children’s Health Defense.
Did Covid Vaccines Really Save Millions?
By Yaakov Ophir | Brownstone Institute | June 21, 2025
Two years have passed since the official end of the Covid-19 pandemic, yet the topic of vaccination remains highly sensitive in both public and scientific discourse. Attempts to question the legitimacy of the mass vaccination campaign or to raise concerns about potential harms are often met with a moral red line: the widely repeated claim that “Covid-19 vaccines have saved millions and millions of lives.”
Remarkably, this assertion was treated as established fact even during the recent U.S. Senate PSI hearing on May 21, 2025, which focused on vaccine-related adverse outcomes.1 Ranking Member Richard Blumenthal opened the hearing with the following statement:
“As we talk about the side effects of COVID vaccines, I think we need to be clear about the most important fact. For all Americans, COVID-19 vaccines have saved millions and millions of lives. There is no scientific question about that fact… One study found that 3 million American deaths were averted…in the United States… I would like this study entered into the record.1
This confident assertion raises a fundamental question: Is there truly solid and conclusive scientific evidence to support the powerful claim that the Covid-19 mass vaccination campaign resulted in a net benefit of millions of lives saved?
Faced with this fundamental question, our research team undertook a structured, step-by-step evaluation of the empirical foundations of the “millions saved” narrative. Building on our prior work,2, 3 we critically examined the hypothetical statistical models that produced this extraordinary figure, as well as multiple randomized controlled trials and large-scale observational studies that served as the empirical basis for the vaccine efficacy estimates fed into these models.
We have now uploaded our full-length article with what we believe to be urgently important findings to a preprint server,4 in order to allow scientists, physicians, and policymakers to independently evaluate the evidence. Because meaningful scientific discourse requires careful scrutiny of the data, we strongly urge readers not to rely solely on the current brief article, but to engage directly with the full analysis presented in our preprint.4
Our goal here is to highlight several central findings that, in our view, demand serious attention, given their direct relevance to one of the most significant public health interventions in modern history: a global, government-backed mass vaccination campaign that, in many countries, was accompanied by mandates and unprecedented restrictions on individual freedoms.
What follows is a concise overview of key insights from our structured analysis that, in our view, every health professional, policymaker, and citizen deserves to consider:
- The widely cited claim that “millions of lives were saved” by Covid-19 vaccines is based on hypothetical models that rest on a long sequence of assumptions—many of which are either weak, unvalidated, or demonstrably false (see below). As a result, the outputs of these models are of questionable value and cannot be taken as reliable evidence.
- A central assumption underlying these models was that Covid-19 vaccines provided strong and durable protection against infection and transmission. Consider the original statement by Dr. Anthony Fauci, then Chief Medical Advisor to the US President: “When you get vaccinated you not only protect your own health… but also you contribute to the community health by preventing the spread of the virus throughout the community…you become a dead end to the virus” (bold added).5 This assumption—serving as the cornerstone of the mass vaccination campaign—turned out to be false. Real-world data quickly revealed that vaccine efficacy against infection was fragile and short-lived, and efficacy against transmission was never directly studied.
- Strikingly, despite the collapse of this original narrative (point 2), the vaccination campaign continued under a revised justification: that the vaccines provide lasting protection against severe illness and death, even after their short-term effect against infection diminishes. It is important to recognize that this updated claim hinges on a conceptual separation between these two types of efficacy—a separation that, as we demonstrate repeatedly in our preprint article, was never empirically validated.
- In fact, available data suggest that protection against infection and protection against severe illness or death are closely linked, following a similar trajectory of waning over time. The difference lies primarily in timing, with a natural delay between initial infection and the development of severe outcomes.
- To directly assess the validity of this supposed distinction between protection against infection and protection against severe illness, we examined the conditional probability of severe illness among individuals who became infected across several key studies. The results were clear: the apparent protection against severe outcomes was most likely a byproduct of the short-term protection against infection. None of the influential studies we analyzed demonstrated independent or durable protection against severe illness or death.
- Notably, some studies stopped tracking severe outcomes precisely at the point when vaccine protection would be expected to wane—paralleling the well-documented decline in protection against infection and the typical delay between infection and the onset of severe illness or death mentioned above. This pattern raises serious concerns about potential misrepresentation or selective reporting of research findings.
- Finally, the pivotal randomized controlled trial that led to the Emergency Use Authorization (EUA) of the Pfizer vaccine showed no meaningful difference between the vaccine and placebo groups in preventing: (1) flu-like symptoms, (2) severe Covid-19, or (3) all-cause mortality. The only significant difference was observed in a non-clinical outcome—laboratory-confirmed Covid-19 infection—and even this result was based on data from no more than 8.24% of participants, collected in a potentially biased manner, as detailed in our preprint.
- Notably, no Covid-19-related deaths were recorded in Pfizer’s pivotal trial. This absence raises serious questions about whether the legal and medical criteria for issuing an emergency use authorization were truly met.
- Even more importantly, the six-month follow-up trial by Pfizer reported 15 deaths in the vaccine group (n = 21,720), compared to 14 in the placebo group (n = 21,728). Given the large sample size, this lack of mortality benefit should have served as a critical anchor for any hypothetical model or evidence-based discussion regarding the overall benefit of the vaccine.
These findings seriously challenge the notion that Covid-19 vaccines saved millions of lives. Moreover, our in-depth investigation uncovered a broader range of methodological flaws that cast doubt on the overall reliability of the existing evidence base. These include: (a) followup periods that were exceedingly short and inconsistently applied across groups; (b) implausible efficacy signals appearing almost immediately after vaccination—well before full immunization could have occurred biologically; and (c) heavy reliance on observational data vulnerable to Healthy Vaccinee Bias, differential testing rates, and numerous other confounding factors.
Taken together, these methodological and empirical concerns not only undermine the foundation of the “millions saved” narrative, but also raise a deeper question: If the evidence is so limited and flawed, how did this narrative gain such dominance in scientific and public discourse?
The issue is not whether some degree of vaccine efficacy was observed at specific moments (e.g., see the fascinating example in our preprint of the Bar-On et al. study on the second booster), but rather how such fleeting observations came to shape the broader public narrative. Isolated data points were elevated and decontextualized, while critical considerations—such as (a) waning immunity, (b) the lack of demonstrated mortality benefit, (c) vaccine breakthrough infections leading to hospitalization or death, and (d) an increasingly robust body of evidence on adverse effects—were systematically sidelined (Figure 1).
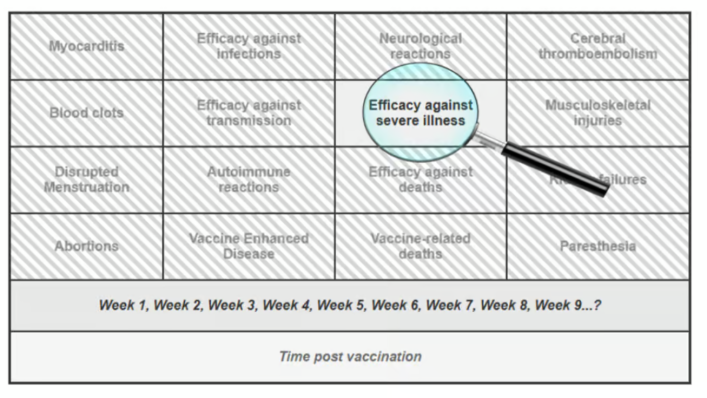
Figure 1. Illustrating a Selective Focus on a Transiently Favorable Outcome While Ignoring Concerning Data
This narrowing of focus — peering through the keyhole of one transient success — has allowed a fragile claim to solidify into a powerful myth, reinforced by institutional authority, social conformity, and the systematic suppression of dissenting voices (including our own experience of censorship, as detailed in our preprint).
We therefore call on the scientific and medical communities to take a step back, widen the lens, and return to a foundational principle of medicine: every intervention, no matter how promising, must undergo continuous, evidence-based evaluation of both its benefits and its potential harms. To the best of our knowledge, such a balanced and rigorous appraisal has yet to be applied to the Covid-19 vaccines.
Based on the evidence reviewed in our preprint, we conclude that the claim that “Covid-19 vaccines saved millions and millions of lives”1 is not supported by empirical evidence. While these vaccines were widely promoted as safe and effective, accumulating reports of serious adverse events—such as myocarditis, pericarditis, thrombosis, and neurological symptoms—have been extensively documented across pharmacovigilance systems and in multiple peer-reviewed studies (e.g., 6-16), many co-authored by the last author of the current article.
Notably, this biologically active intervention was administered repeatedly in the form of boosters, thereby compounding potential risks—often in populations with near-zero risk of Covid-related mortality, such as children. Taken together with the lack of demonstrable long-term efficacy presented in our preprint,4 the available evidence suggests that the risk–benefit balance of the Covid-19 vaccines may, in fact, tilt toward the negative end of this fundamental medical equation.17, 18
References
1. Homeland Security. The Corruption of Science and Federal Health Agencies: How Health Officials Downplayed and Hid Myocarditis and Other Adverse Events Associated with the COVID-19 Vaccines.
2. Ophir Y, Shir-Raz Y, Zakov S, McCullough PA. The Efficacy of COVID-19 Vaccine Boosters against Severe Illness and Deaths: Scientific Fact or Wishful Myth?. Journal of American Physicians and Surgeons. 2023;28(1). doi: https://www.jpands.org/vol28no1/ophir.pdf.
3. Ophir Y. The Final Brick in the Vaccine Efficacy Narrative ⋆ Brownstone Institute. 2023.
4. Ophir Y, Shir-Raz Y, Zakov S, McCullough PA. A Step-by-Step Evaluation of the Claim That COVID-19 Vaccines Saved Millions of Lives. Researchgate (preprint). 2025. doi: 10.13140/RG.2.2.12897.42085.
5. NEWS C. Transcript: Dr. Anthony Fauci on “Face the Nation,” May 16, 2021. 2021.
6. Rose J. A Report on the US Vaccine Adverse Events Reporting System (VAERS) of the COVID-1 9 Messenger Ribonucleic Acid (mRNA) Biologicals. Science, Public Health Policy, and The Law. 2021;2:59–80.
7. Fraiman J, Erviti J, Jones M, et al. Serious adverse events of special interest following mRNA COVID-19 vaccination in randomized trials in adults. Vaccine. 2022;40(40):5798–5805. doi: 10.1016/j.vaccine.2022.08.036.
8. Shir-Raz Y. Breaking: Leaked Video Reveals Serious Side-Effects Related to the Pfizer COVID-19 Vaccine Covered Up by the Israeli MOH. 2022.
9. Witberg G, Barda N, Hoss S, et al. Myocarditis after Covid-19 Vaccination in a Large Health Care Organization. N Engl J Med. 2021;385(23):2132–2139. doi: 10.1056/NEJMoa2110737.
10. Chua GT, Kwan MYW, Chui CSL, et al. Epidemiology of Acute Myocarditis/Pericarditis in Hong Kong Adolescents Following Comirnaty Vaccination. Clinical Infectious Diseases. 2021:ciab989. doi: 10.1093/cid/ciab989.
11. Hulscher N, Alexander PE, Amerling R, et al. A Systematic REVIEW of Autopsy findings in deaths after covid-19 vaccination. Forensic Sci Int. 2024:112115. doi: 10.1016/j.forsciint.2024.112115.
12. Oster ME, Shay DK, Su JR, et al. Myocarditis Cases Reported After mRNA-Based COVID-19 Vaccination in the US From December 2020 to August 2021. JAMA. 2022;327(4):331–340. doi: 10.1001/jama.2021.24110.
13. Takada K, Taguchi K, Samura M, et al. SARS-CoV-2 mRNA vaccine-related myocarditis and pericarditis: An analysis of the Japanese Adverse Drug Event Report database. Journal of Infection and Chemotherapy. 2024.
14. McCullough P, Rogers C, Cosgrove K, et al. Association between COVID-19 Vaccination and Neuropsychiatric Conditions. 2025.
15. McCullough PA, Hulscher N. Risk stratification for future cardiac arrest after COVID-19 vaccination. World J Cardiol. 2025;17(2):103909. doi: 10.4330/wjc.v17.i2.103909.
16. Hulscher N, Hodkinson R, Makis W, McCullough PA. Autopsy findings in cases of fatal COVID-19 vaccine-induced myocarditis. ESC Heart Failure. 2024;n/a. doi: 10.1002/ehf2.14680.
17. Mead MN, Seneff S, Wolfinger R, et al. COVID-19 Modified mRNA “Vaccines”: Lessons Learned from Clinical Trials, Mass Vaccination, and the Bio-Pharmaceutical Complex, Part 1. International Journal of Vaccine Theory, Practice, and Research. 2024;3(2):1112–1178. doi: 10.56098/fdrasy50.
18. Mead MN, Seneff S, Rose J, Wolfinger R, Hulscher N, McCullough PA. COVID-19 Modified mRNA “Vaccines”: Lessons Learned from Clinical Trials, Mass Vaccination, and the Bio-Pharmaceutical Complex, Part 2. International Journal of Vaccine Theory, Practice, and Research. 2024;3(2):1275–1344. doi: 10.56098/w66wjg87.
This article was co-authored by Yaffa Shir-Raz, Shay Zakov, and Peter A. McCullough.
Dr. Yaakov Ophir is Head of the Mental Health Innovation and Ethics Lab at Ariel University and a member of the Steering Committee for the Centre for Human-Inspired Artificial Intelligence (CHIA) at the University of Cambridge. His research explores digital-age psychopathology, AI and VR screening and interventions, and critical psychiatry. His recent book, ADHD Is Not an Illness and Ritalin Is Not a Cure, challenges the dominant biomedical paradigm in psychiatry. As part of his broader commitment to responsible innovation and scientific integrity, Dr. Ophir critically assesses scientific studies related to mental health and medical practice, with particular attention to ethical concerns and the influence of industrial interests. He is also a licensed clinical psychologist specializing in child and family therapy.
MenQuadfi Approval and the Pyramid Scheme of Vaccine Safety
This is how the game is played
Injecting Freedom by Aaron Siri | June 15, 2025
Recently, FDA shamefully approved Sanofi’s MenQuadfi (a meningococcal vaccine) to be injected into infants 6 weeks to 2 years old based on a trial that compared it to Menveo (another meningococcal vaccine). In the trial, 5.3% of infants receiving MenQuadfi and 3.6% of infants receiving Menveo had a serious adverse reaction (which means something very serious, see the FDA definition). But because these rates were “similar,” this product was deemed “safe” by FDA—because it assumes Menveo is also “safe.”
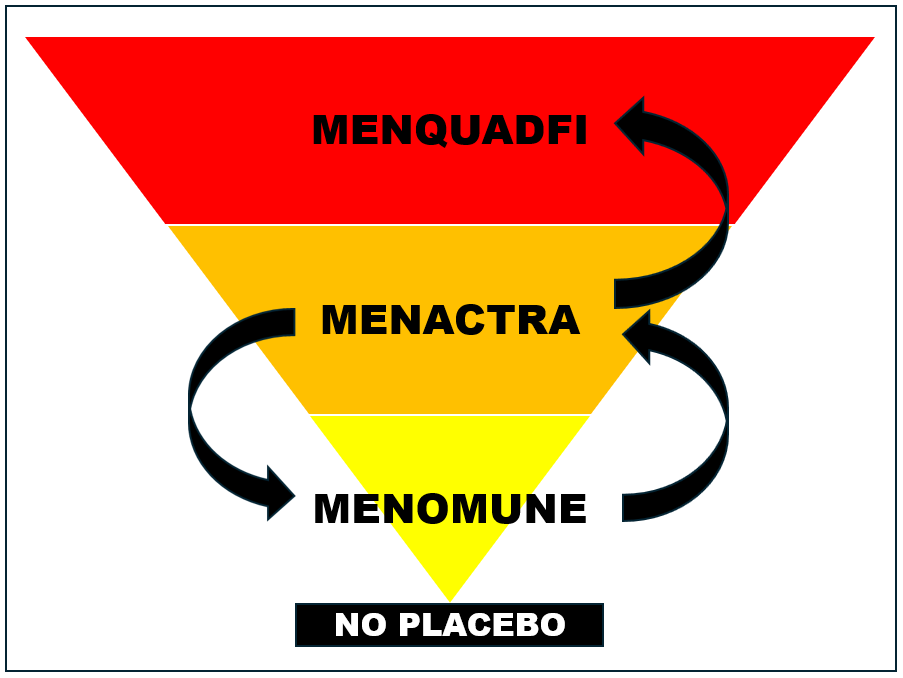
But Menveo was licensed based on a trial in which Menactra (among other vaccines) was used as a control; and Menactra was licensed based on a trial in which Menomune was used as a control; and Menomune was not licensed based on a proper placebo-controlled trial either. In fact—and this is mind-twisting—the package insert for Menomune lists the clinical trial for Menactra (in which Menomune itself was used as the control) as the basis for its safety. I couldn’t even dream of making this stuff up.
This provides a good example of the vaccine safety pyramid scheme: Menomune was licensed without a proper placebo-controlled trial and was then used as the control to license Menactra; Menactra is then used as the control to license Menveo; and then Menveo is used as the control to license MenQuadfi. And then we get a trial with 5.3% and 3.6% of infants suffering serious adverse reactions and FDA grants licensure.
What makes this even more troubling is that because FDA officials must know these numbers are highly concerning, they have Sanofi conduct a case-by-case review of each serious adverse event. If FDA were confident the control was safe, it would just rely on a statistical comparison between the vaccine being evaluated and the control. But since FDA officials must know Menveo’s safety is unknown (because it was licensed in a trial with a defective control), they ask Sanofi (the company seeking to get approval and profit from this product) to explain away each serious adverse event. And the Sanofi-paid researchers do exactly that in their write-ups to FDA about each serious adverse event.
And when FDA gets these write-ups explaining away each serious adverse event as “unrelated” to MenQuadfi, what does FDA do? The FDA officials reviewing them dutifully agree. What else would they do? Admit that Menveo, used as the control, and which they licensed based on nonsense data, has been harming children? That it is causing 3.6% of children—or even a fraction of that—to have a serious adverse event? If FDA officials do that, the house of cards would start to collapse. It would become clear Menveo wasn’t properly licensed (which it wasn’t), and that Menactra wasn’t properly licensed (which it wasn’t), and the same for Menomune.
FDA’s conflict and bias are dangerous. Letting Sanofi decide if its own product caused harm is beyond dangerous. This entire pyramid scheme, without a valid baseline of safety permitting a statistical comparison, requires injecting a new layer of biased assumptions with each additional licensure.
At this point, the safety of these products is based on dogma and assumptions. FDA has its reputation and any remaining trust to lose—and pharma its billions in profits—if they actually evaluated these products using a true safety comparator. It would reveal the true safety profile of these products (which the reliable data shows will likely be terrifying). Of course, the children whose injuries could be averted by conducting actual safety trials would benefit, but they are not really part of the equation.
References:
Public Ridicule Hyped Summer Heat Headlines… Meteorologists Losing Credibility
Sensationalist weathermen in Germany losing credibility, get mocked and ridiculed.
By P Gosselin | No Tricks Zone | June 18, 2025
Meteorologists are discovering that if they want to get attention from the media and more clicks and and likes (short term), then all they have to do is announce fictional heatwaves that weather models routinely hallucinate 10-14 days out. “Temperatures could soar to 40°C!”
German online Weltwoche reports on this phenomenon with a recent article titled: “The “heatwave” to be followed by the “red hot wave”: the climate alarmists are taking themselves to the point of absurdity and losing all credibility.”
Legacy media has a meltdown after RFK Jr fires the CDC’s vaccine panel
By Maryanne Demasi, PhD | June 10, 2025
Yesterday, Robert F. Kennedy Jr. fired every single member of the CDC’s Advisory Committee on Immunization Practices (ACIP)—the influential group of experts that decides which vaccines are added to the childhood schedule.
Today, he set fire to the media’s hysterical reaction.
Within 24 hours, legacy outlets and public health institutions lost their collective minds. Former CDC directors, industry-funded doctors, and conflicted public health groups lined up to denounce Kennedy’s move as reckless, anti-scientific, even deadly.
“This is a dangerous and unprecedented action that makes our families less safe,” said former CDC director Dr Tom Frieden.
“Unilaterally removing the entire panel of experts is reckless,” said paediatrician Dr Tina Tan to The New York Times.
The American Academy of Pediatrics (AAP) said it was “deeply troubled and alarmed.” It claimed the move would “stoke distrust in lifesaving vaccines”—this from the same organisation that has spent decades pushing the childhood vaccine schedule while taking money from the very companies that profit from it.
Others framed it as a political purge, a blow to science, or a “coup” that would bring back measles and polio.
But within hours, Kennedy hit back—and this time, he wasn’t the outsider being easily dismissed. He was the Secretary of Health and Human Services. And he came armed with evidence, receipts, and a brutal takedown of the media’s favourite falsehoods.
In a searing post on X, Kennedy explained the decision.

He said the clean sweep was necessary because ACIP had demonstrated its “stubborn unwillingness to demand adequate safety trials before recommending new vaccines for our children.”
And despite the media’s insistence otherwise, Kennedy argued that no routine injected childhood vaccine on the CDC’s schedule had ever been approved based on a placebo-controlled trial using an inert substance.
CNN had tried to prove him wrong last week—claiming it had found “257 placebo-controlled studies” of vaccines on the schedule.
Kennedy dismantled it in forensic detail.
“CNN is wrong,” he wrote. “No routine injected vaccine on CDC’s schedule was licensed for children based on a placebo-controlled trial. That is not conjecture. It is a fact based on FDA’s clinical trial data.”
Then came the body blows.
He pointed out that most of the 257 studies used active substances like aluminium, antibiotics, or other vaccines—not inert placebos.
He linked directly to FDA definitions of “placebo” and to official clinical trial records. Of the few studies that may have used saline controls, none were relied on by the FDA to license a single routine vaccine for American children.
Some studied products that were never approved in the US. Some occurred after licensure. Others involved discontinued vaccines. “CNN’s list ironically proves the lack of adequate safety trials,” Kennedy wrote in a stinging rebuke.
The post was devastating.
It was a clinical takedown of an industry riddled with deception—and it landed—because this time, Kennedy wasn’t being filtered through a hostile press.
He was speaking directly to the public, as a government official, with all the links to back it up. And the media couldn’t handle it.
Predictably, the media rolled out the same tired “experts” to recycle the same tired script—Paul Offit quotes, panic about “undermining trust,” warnings that children would die.
But Kennedy turned the whole thing inside out.
“We’ve gone from three routine injections by age one in 1986 to 25 in 2025,” he wrote. “And not one of them was licensed using a placebo-controlled trial.”
He said it plainly for the cameras: “That is just malpractice. So the people who are in charge of that are now gone.”
For years, the press had written Kennedy off as an anti-vaxxer and moved on. Now, they’ve thrown everything at him—and he threw it right back. Only now, he has the authority, data, and reach.
Kennedy told his followers he’d be announcing replacements in the coming days—no “ideological anti-vaxxers” just “highly credentialed physicians and scientists” committed to evidence, objectivity, and common sense.
Legacy media may still control the headlines, but they can no longer suppress the debate.
And perhaps that’s what really has them rattled.
They’re not defending science. They’re defending a regime of experts who signed off on decades of vaccine approvals without ever insisting on rigorous, inert-placebo safety trials.
When Kennedy calls them out, their only defence is to scream “danger!”—and hope no one checks the fine print.
Yesterday, he fired the gatekeepers. Today, he exposed the game.
Featured Video

or go to
Aletho News Archives – Video-Images
From the Archives
Israel Would Have No Qualms About USS Liberty-Style FALSE FLAG If Iran Campaign Falters – Analysts
By Ilya Tsukanov – Sputnik – 18.06.2025
Donald Trump is mulling whether or not to join Israel’s aggression against Iran as Tel Aviv faces problems sustaining its defenses against growing counterstrikes, and apparently lacks a realistic game plan for an end to hostilities after failing to achieve its goals. Analysts told Sputnik how the US could be ‘nudged’ into the conflict.
“The US is already assisting Israel with supplies, intel, refueling support, etc. One of the many US posts in the region could be attacked for a casus belli,” former Pentagon analyst Karen Kwiatkowski explained.
“If Trump doesn’t comply with Israel’s demand” and join its aggression voluntarily, “a false flag may be needed” to drag the US in, Kwiatkowski, retired US Air Force Lt. Col.-turned Iraq War whistleblower, fears.
Netanyahu has a diverse array of options at his disposal, according to the observer, including:
- a false flag against US assets abroad blamed on Iran or one of its Axis of Resistance allies, like the Houthis
- a US domestic attack or assassination blamed on Iran
- Iranian air defenses ‘accidentally’ hitting a civilian jetliner carrying Americans
- use of a dirty bomb or nuclear contamination somewhere in the region blamed on Iran
- even blackmailing by threatening to use nukes against Iran if the US doesn’t join the fight
Kwiatkowski estimates that Israel probably has “enough blackmail power” against President Trump and Congress to avoid the necessity of a false flag operation, but a “USS Liberty-style” attack, targeting the soon-to-be-retired USS Nimitz supercarrier that’s heading to the Middle East, for example, nevertheless cannot be ruled out entirely, she says. … continue
Blog Roll
 Aletho News
Aletho News- Embarrassing Pivot: U.S. considers dropping Iran oil sanctions amid war
- US has 2 months of rare earths left to replenish weapons amid Iran war: Report
- Strait Of Hormuz closure brings Empire to brink
- Iran controls Strait of Hormuz, dictates terms of war and peace as US excursion backfires
- Outlasting the Superpower: The Reasons Why Iran is Beating uhe US and Israel
- Top PMU commander, over a dozen fighters killed in new US strikes on Iraq
- Is Netanyahu’s war gamble threatening the future of ‘Israel’?: FT
- Smotrich calls for annexation of South Lebanon to Litani River
- Echos of Gallipoli? Hormuz and the Geography of Hubris
- Pakistan PM backs Iran’s right to self-defense amid tensions
- If Americans Knew
- UN’s special rapporteur on human rights says Israel is systematically torturing Palestinians
- Trump White House plagiarized Iran war manifesto from Israel-aligned think tank
- Gaza says 6–10 patients die daily waiting for treatment abroad as Israel blocks medical evacuations
- ‘Substantial evidence’ of double-tap strike in killing of Gaza’s Hind Rajab
- ‘Do Not Want To Die For Israel’: Doubts About Trump’s Iran Strategy Spread Among Troops
- Instead of taking Joe Kent’s claims seriously, the media is disregarding him as an antisemite
- Joe Kent’s Explosive Interviews about his Resignation over the Iran War
- Israel poised to take over southern Lebanon; settlers wreak havoc in West Bank – Not a ceasefire Day 165
- From Sde Teiman, the truth about Israel’s military justice system has been set free
- Child denied life-saving bone marrow transplant by Israel ‘because he is from Gaza’
- No Tricks Zone
- Devastating Assessment Of Comirnaty Vaccine By Former Senior Pfizer Europe Toxicologist
- New Study: CO2 Is ‘Effectively Negligible’ As An Explanatory Climate Change Factor Since 2000
- Former Pfizer Toxicologist Dr. Helmut Sterz Tells Bundestag Hearing Pfizer Vaccine Should Have Never Been Approved
- Energy Expert: Germany’s Nuclear Phaseout Was A “500 Billion Euro Mistake”
- New Research: South Australia’s Mid-Holocene Sea Surface Temperatures Were 4°C Warmer Than Today
- Storing Green Energy To Last Germany 10 Days Would Require A 60-Million Tonne Battery
- New Studies: UK Sea Levels Were 4 Meters Higher Than Today During The Mid-Holocene
- Destructive Green New Deal: German Energy And Metal Group Warns Of Drastic Crisis
- New Study Documents A 20-Year Pause In Arctic Sea Ice Decline – Driven By Internal Variability
- Wake-up Call: Survey Shows Majority Of Germans Now Favor Postponing Climate Targets!
