Critics of failed COVID policies call for pandemic reckoning
By Daniel Nuccio | Washington Examiner | February 15, 2023
Eight leading critics of the United States’s COVID-19 response have called for an investigation of the many failures of policy architects and key decision makers — at institutions ranging from the Centers for Disease Control and Prevention and Food and Drug Administration to universities and hospitals — over their repeated mishandling of the pandemic.
Given the immense harm inflicted on our society by the follies of a ruling class and their expert advisers who never failed to make a wrong decision when presented with the opportunity, as well as the fact that lives are still being destroyed by their lingering policies, we can only hope this blueprint does not go ignored.
Dubbing themselves the “Norfolk Group,” the association of scholars includes such prominent names as Stanford epidemiologist Jay Bhattacharya, Harvard epidemiologist Martin Kulldorff, UCSF physician Tracy Beth Høeg, Johns Hopkins University surgeon Marty Makary, and Indiana University School of Medicine immunologist Steven Templeton.
According to the Norfolk Group’s website, although initially organized by the Brownstone Institute in May 2022, the eight members of the group have since worked free from outside influence to draft the 80-page document they published earlier this year, “Questions for a COVID-19 Commission.”
Presented as a series of summaries and questions pertaining to key elements of U.S. COVID policy, the document, in effect, lays out a thorough indictment of the consistent incompetence of our ruling class while also raising concerns over the possible influence on policy by special interests such as teachers unions and drug companies.
Regarding natural immunity, the authors ask, “Why did the CDC downplay infection-acquired immunity, despite robust evidence for it?”
In respect to school closures, they ask, “Why were schools and universities closed despite early evidence about the enormous age-gradient in COVID-19 mortality … and early evidence that school closures would cause enormous collateral damage to the education and mental health of children and young adults?”
On that matter, they also wonder, “Why did the CDC incorporate policy language proposed by leaders of teachers unions on the scientific and public health aspects of school reopening without soliciting expertise of outside scientists in public health, infectious diseases, or other related fields?”
When discussing lockdowns, they inquire, “Why was so much influence on public health policy accorded to Drs. [Francis] Collins and [Anthony] Fauci? They control the largest source of infectious disease research funding in the world. How many infectious disease scientists, who should have been strong voices during the pandemic, kept quiet for fear of losing the research funding on which their livelihood depends?”
In their section on epidemiologic modeling, they demand, “Why did world leaders overly rely on models that made unverified assumptions about the pandemic’s trajectory rather than trying to verify these assumptions and their implications?”
When addressing COVID-19 vaccines, they raise questions such as, “Why did many organizations continue with mandates through summer and fall of 2021, despite data demonstrating both waning efficacy of symptomatic infection and reduced long term ability to curb viral spread?”
Regarding masks, they state, “Prior to the COVID-19 pandemic, the evidence that masks did little if anything to stop the spread of respiratory viruses was uncontroversial,” before summarizing a few studies demonstrating this and asking the obvious: “[W]hy did public health officials and agencies promote the idea that masks would be effective against SARS-CoV2?”
In its entirety, the Norfolk Group’s “Questions for a COVID-19 Commission” serves as a blueprint for the kind of investigation our country needs. Just don’t expect the Biden administration to do anything about it.
Daniel Nuccio holds master’s degrees in both psychology and biology. Currently, he is pursuing a PhD in biology at Northern Illinois University studying host-microbe relationships. He is also a regular contributor to The College Fix where he writes about COVID, mental health, and other topics.
Is the FDA “doubling down” on a failed strategy?
By Maryanne Demasi, PhD | January 30, 2023
Last Thursday, the FDA convened its Vaccines and Related Biological Products Advisory Committee (VRBPAC) to discuss the future of covid vaccines.
The panel voted 21 to 0 in favour of moving towards a more simplified vaccine schedule – an annual shot which would be updated as new variants emerge – much like the annual flu shot.
Despite the unanimous vote, VRBPAC members did raise concerns about knowledge gaps and questioned the need to boost everyone, as well as the futility of chasing rapidly mutating viruses.
But it all fizzled out quickly, and the FDA promised to reconvene in May or June to discuss the data further.
That said, I had some interesting observations of my own.
We are three years into the pandemic, and the FDA has still not established a “correlate of protection” for the vaccines.
Eight covid-19 vaccine emergency use authorisations (EUAs)* have been granted, based on their ability to induce “neutralising antibodies,” a surrogate marker of protection.
The idea is, the more antibodies you produce, the better you are protected.
Except, neutralising antibodies do not predict the degree to which someone is protected from infection… and the FDA knows it.
Ofer Levy, VRBPAC member and Professor of Paediatrics at Boston Children’s Hospital first voiced his concern at the April 6, 2022 meeting.
“We’re at risk of doubling down on a failed strategy,” said Levy as the committee discussed a framework for offering annual covid shots for Americans.
“Where is the federal effort to coordinate all of that to develop a public repository around the correlate of protection, and to make sure we have the best available data for the immunogenicity when we make those decisions?”
The FDA’s top vaccine official, Peter Marks, agreed with Levy.
“There is not a clear, perfect, immune correlate of protection” admitted Marks, “We’re using poor man’s immune correlates of protection here — or poor person’s immune correlates of protection with antibody levels.”
In Dec 2022, Peter Marks reiterated these concerns in an article published in JAMA. He and his co-authors wrote:
“Therefore, unless correlates of protection that are strongly associated with duration of protection against COVID-19 can be identified, it is likely that rather than relying on immunobridging to infer vaccine effectiveness, large randomized clinical trials similar to the initial trials of the currently authorized or licensed vaccines for COVID-19 will be required to ascertain the effectiveness of these new vaccines.”
But fast forward to this latest meeting, and it becomes clear that we’re all still in the dark.
We have no correlate of protection, the FDA is relying heavily on real world studies (confounded data) and the agency still has not demanded any randomised controlled trials to show the bivalent booster can reduce severe disease or hospitalisations.
It’s no wonder doctors are coming out in droves, refusing to have any more covid shots until the FDA demands better studies.
“I don’t think we can say with credibility what the objective benefits are for someone like me to take an additional dose, nor what the rate of any rare but important side effects would be,” tweeted Todd Lee, a physician certified in Infectious Diseases and General Internal Medicine in Quebec, Canada.
Similarly, Vinay Prasad, haematologist-oncologist at the University of California San Francisco vowed not to take any more shots until there were data from randomised controlled trials.
“I took at least 1 dose against my will. It was unethical and scientifically bankrupt. I am not done with that error. No more,” he tweeted.
As part of its post-marketing requirements, Pfizer is legally obligated to conduct a study involving people aged 16 to 30 to look at rates of subclinical myocarditis (i.e. underlying damage to the heart muscle without causing symptoms).
The final report was due 31 Dec 2022, but that deadline lapsed, and the FDA said nothing. There was no mention of the study, neither in the briefing notes ahead of the VRBPAC meeting, or during the meeting.
I asked the FDA directly for access to Pfizer’s study, but the agency said in an email, “You may submit a FOIA request for this information, or if you would like it more quickly, you can reach out to the manufacturer directly.”
Pfizer did not respond to my request, and the FDA refused to confirm whether it had even received Pfizer’s study, before abruptly ending our communication.
Jessica Adams, an expert in drug regulatory affairs pointed out on twitter that the FDA had quietly changed the due date for the study from 31 Dec 2022 to 30 June 2023.

So, now as it stands, millions of young people will receive boosters, mandated or not, without knowing if the vaccine is causing subclinical myocarditis.
FDA still working from home
Finally, the meeting was again held online because the majority of FDA employees are still working from home.
Since all federal employees have been mandated to take the covid-19 vaccine to “protect themselves and those around them,” why aren’t they conducting face-to-face meetings?
“FDA leaders are in a bubble. How much longer will the FDA (18,000-employees) continue to work remotely? It’s mid-day on a weekday and the parking lot is essentially empty” tweeted Marty Makary, surgeon and public policy researcher at Johns Hopkins University.
“The FDA was telling the rest of America to get vaccinated, mask up and go back to work, but the FDA mysteriously did not follow its own advice,” said David Gortler, drug safety expert and former senior advisor to the FDA commissioner.
Well, it’s as though the FDA heard the cries.
Today, the FDA announced that “staff will be transitioning to a hybrid workplace.” This transition will enable face-to-face formal meetings between FDA and industry to resume within weeks.
*FDA issued eight EUAs based on neutralising antibodies (immunobridging studies) – an unproven correlate of protection.
- Pfizer EUA – 6 months to 4 year olds
- Pfizer EUA – 5 to 11 year olds
- Pfizer EUA – 12 to 15 year olds
- Pfizer EUA booster #1
- Pfizer EUA booster #2
- Moderna EUA – 6 months to 17 year olds
- Moderna EUA – booster #1
- Moderna EUA – booster #2
Drink it, snort it, smoke it – the vaccine juggernaut rumbles on
By Roger Watson | TCW Defending Freedom | January 28, 2023
More good news on vaccine, folks. First, you may be required to take only one Covid-19 shot per year, and if all goes well you will not even have to do that. You will be able to drink or even inhale your vaccine. No more painful injections, just a quick slurp or a snort and the job’s a good ’un. That’s you safe from the deadly virus for another year.
We could even make it fun. Why not hold Covid-19 vaccine parties? A selection of flavours in shot glasses (they don’t call them shot glasses for no reason) or add your vaccine to a vape and puff away until your immune system is primed.
I glean all this garbage from Global Health Now, the daily newsletter from the Johns Hopkins Bloomberg School of Public Health. The first story concerns how the Food and Drug Administration (FDA) in the United States is considering ‘simplifying the Covid vaccination schedule, allowing most people to get the currently available booster, regardless of how many doses they had received before that’. This means that if you are boosted up to the eyeballs or have never had one before and suddenly made the incomprehensible decision to start now, then Bob’s your uncle; roll up your sleeves.
Please note that nothing has changed; there is no new vaccine and no new threat. The FDA is just making an arbitrary decision to change the schedule. Clearly the aim is to get more people to accept the vaccination. But it is also clear that they are making this stuff up as they go along. They have no further evidence that the vaccines will work any better this way.
The information that is available to them is the abundant and accumulating evidence of vaccine harms which, incredibly, the Medicines and Healthcare products Regulatory Agency (MRHA) in the United Kingdom admits can be serious while insisting that the vaccines are safe. If truth is the first casualty of war – it certainly died early in the Covid-19 madness – logic is not far behind it. The MRHA is willing to trade off serious vaccine side effects against minimal protection from a virus which is virtually harmless to the vast majority of people. Perhaps the FDA is trying to reduce the number of boosters it says people will need in the hope that vaccine injuries will go away. Alternatively, it may be keen to accelerate the rollout before the general population wakes up to the fact that they are being conned, if they are lucky, and killed if they are not.
The potential for a drinkable/snortable/inhalable vaccine comes courtesy of US Speciality Formulations, a company which has produced the QYNDR vaccine. If QYNDR is a bit of a consonant-rich mouthful, then be informed that the official pronunciation if ‘KINDER’. And the advent of QYNDR is closer than you think. Phase 1 trials have already been completed in New Zealand (where else?) and all that is required is more funding to proceed with further trials. Apparently, it is very difficult to formulate a vaccine that survives the vicissitudes of the digestive tract.
And why do we need these vaccines? Well, according to US Speciality Formulations: ‘Covid-19 is still here and deadly.’ Also, I imagine that the inventors and investors envisage that this will make them shedloads of money. It clearly pays to perpetuate the Covid-19 narrative and to pepper it with as much panic as possible.
At some point in the panic-demic, the vaccine rollout became a juggernaut. Large and hard to stop. With the widespread and obvious extent to which people are gullible, government and drug manufacturers are willing to lie, health professionals are willing to stay silent and there are bucks to be made, it is unlikely that the juggernaut will be halted any time soon.
Who knows what’s next? Perhaps they will develop a vaccine that one can stick up one’s bottom. Whether or not they do, I strongly advise them that is what they can do with the present products.
Merck’s Taxpayer-Subsidized COVID Pill Linked to New Virus Mutations, Study Finds
By Michael Nevradakis, Ph.D. | The Defender | February 3, 2023
Merck’s oral antiviral pill for COVID-19, molnupiravir — marketed under the name Lagevrio — may be fueling the development of new and potentially deadly variants of COVID-19, according to the authors of a new preprint study.
The study, released Jan. 27 by a team of U.S. and U.K researchers, found, “It is possible that some patients treated with molnupiravir might not fully clear SARS-CoV-2 infections, with the potential for onward transmission of molnupiravir-mutated viruses.”
Merck received significant taxpayer funding from the Biden administration to develop and distribute molnupiravir, and the U.S. government bought nearly 2 million courses of the drug on the taxpayer’s dime.
The study, which is pending peer review, followed the discovery by a middle school science and math teacher in Indiana who found numerous variants of COVID-19 emerged after molnupiravir began to be widely distributed.
Scientists had long warned that the development of such mutations from the use of molnupiravir was possible.
“It’s not a surprise that molnupiravir could cause [the] escape of mutant virus strains or substrains into the population,” said Dr. Harvey Risch. “Its main function is to get the virus to mutate faster.”
Risch, professor emeritus and senior research scientist in epidemiology (chronic diseases) at the Yale School of Public Health, told The Defender :
“The idea is that it will mutate itself to death. But some live mutants could get out, and this paper gives evidence that they have.”
Brian Hooker, Ph.D., P.E., chief scientific officer for Children’s Health Defense, said the study’s authors scanned global SARS-CoV-2 sequence databases looking for mutations characteristic of those by molnupiravir (G-to-A and C-to-U) and found an uptick of those mutants starting in 2022 — after molnupiravir was put on the market and specifically in countries where molnupiravir was distributed.
“Although this isn’t ‘direct proof’ that the mutations came directly from molnupiravir use,” Hooker told The Defender, “the evidence is very compelling, confirming the fears of many who warned of this prior to FDA [U.S. Food and Drug Administration] approval of the drug in late 2021.”
The FDA granted molnupiravir Emergency Use Authorization (EUA) on Dec. 23, 2021, for use in mild-to-moderate COVID-19 infections in patients 18 and over.
The EUA came just one day after the FDA authorized Pfizer’s COVID-19 antiviral treatment Paxlovid.
Merck this week announced massive revenues from sales of molnupiravir in 2022, but projected a significant decrease in those sales in 2023.
The FDA on Wednesday removed the requirement that a person has to test positive for COVID-19 in order to get a prescription for molnupiravir or Paxlovid.
‘I think we are courting disaster’
Molnupiravir “works by creating mutations in the COVID-19 genome that prevent the virus from replicating in the body, reducing the chances it will cause severe illness,” according to Bloomberg.
However, according to Science, the findings of the preprint study suggest “some people treated with the drug generate novel viruses that not only remain viable, but spread.”
This finding “underscores the risk of trying to intentionally alter the pathogen’s genetic code,” leading some researchers to “worry the drug may create more contagious or health-threatening variations of COVID,” Bloomberg reported.
Virologist William Haseltine, Ph.D., chair and president of ACCESS Health International, has repeatedly raised such concerns about molnupiravir.
“It’s very clear that viable mutant viruses can survive [molnupiravir treatment] and compete [with existing variants],” Haseltine told Science. “I think we are courting disaster.”
According to the Gateway Pundit, “When one studies how Lagevrio works, this should not come as a shock. The pill attacks the COVID virus by trying to alter its genetic code.”
The Gateway Pundit reported:
“Once inside a human cell, a virus can make 10,000 copies of its genetic code in a few hours. Each copy made increases the risk the virus makes a rare mistake and creates an inexact replica.
“This is how mutations happen as we have seen with COVID. A drug that deliberately alters a virus’s genetic code would greatly increase the mutation risk.”
Dr. Jonathan Li, a virologist and the director of Li Laboratory, associated with Harvard Medical School and Brigham and Women’s Hospital, told Bloomberg :
“There’s always been this underlying concern that it could contribute to a problem generating new variants. This has largely been hypothetical, but this preprint validates a lot of those concerns.”
According to Science, Haseltine and other scientists have long worried that molnupiravir would create COVID-19 mutations that “would survive and propagate — and perhaps turn out to be more transmissible or virulent than before.”
A Merck spokesperson described that theory as “an interesting hypothetical concern,” prior to the drug receiving EUA.
The same scientists also worried that aside from the virus, the DNA of those receiving the drug might also mutate, Science reported.
These concerns led “researchers and citizen scientists” to examine COVID-19 genome sequences cataloged in the international GISAID (Global Initiative on Sharing Avian Influenza Data) database, seeking to identify mutations likely to be caused by molnupiravir.
‘Clearly something is happening here’
Searching for these mutations was based on the premise that, “Rather than inducing random changes in the virus’ RNA genome, [molnupiravir] is more likely to cause specific nucleic acid substitutions, with guanine switching to adenine and cytosine to uracil,” added Science.
Through this process, Ryan Hisner, a middle school science and math teacher from Monroe, Indiana — described by Science as a “virus hunter” — ultimately “identified dozens of sequences that showed clusters of those hallmark substitutions.”
Hisner took to Twitter with his concerns, where he came into contact with Thomas Peacock, Ph.D., a virologist at the Imperial College London. They and other U.K. and U.S. researchers “systematically reviewed more than 13 million SARS-CoV-2 sequences in GISAID and analyzed those with clusters of more than 20 mutations,” according to Science.
The team found “a large subset showed the hallmark substitutions; all dated from 2022, after molnupiravir began to be widely used,” Science reported.
According to the preprint study, Molnupiravir, “acts by inducing mutations in the virus genome during replication. Most random mutations are likely to be deleterious to the virus, and many will be lethal.”
However, the researchers wrote:
“It is possible that some patients treated with molnupiravir might not fully clear SARS-CoV-2 infections, with the potential for onward transmission of molnupiravir-mutated viruses.
“We set out to systematically investigate global sequencing databases for a signature of molnupiravir mutagenesis. We find that a specific class of long phylogenetic branches appear almost exclusively in sequences from 2022, after the introduction of molnupiravir treatment, and in countries and age groups with widespread usage of the drug.
“Our data suggest a signature of molnupiravir mutagenesis can be seen in global sequencing databases, in some cases with onwards transmission.”
Peacock told Science these “signature clusters” were up to 100 times more likely to be identified in countries where molnupiravir was widely used, including the U.S., U.K. and Australia, as compared to countries such as Canada and France, where it was not in widespread use.
“Clearly something is happening here,” said Peacock.
Merck: ‘no evidence’ any antiviral agent has contributed to the emergence of circulating variants’
Theo Sanderson, Ph.D., a geneticist at the Francis Crick Institute and co-author of the preprint, told Science “We are not coming to a conclusion about risk” just yet, with regard to whether or not these mutations may lead to more severe COVID-19 variants.
Indeed, according to the preprint study, the variants identified by the researchers have not been shown to be more lethal or more evasive to immunity than other existing strains of COVID-19.
However, Haseltine illustrated the potential risk via the analogy of owning a pet lion: “Just because it didn’t bite you yesterday doesn’t mean it won’t bite you today.”
According to the Gateway Pundit :
“Merck was warned by multiple scientists their drug might create problematic mutations which would render the virus more dangerous and difficult to treat. The company decided to blow off any concerns and put Lagevrio [molnupiravir] on the market anyway.”
As previously reported by The Defender, Dr. James Hildreth, president and CEO of Meharry Medical College and member of Biden’s COVID-19 Health Equity Task Force, expressed concerns about mutant variants escaping.
In 2021, Hildreth told an FDA advisory panel, “Even if the probability is very low, one in 10,000 or 100,000, that this drug would induce an escape mutant from which the vaccines we have do not cover, that could be catastrophic for the whole world actually.”
Also in 2021, Haseltine told Science :
“You are putting a drug into circulation that is a potent mutagen at a time when we are deeply concerned about new variants. I can’t imagine doing anything more dangerous.
“If I were trying to create a new and more dangerous virus in humans, I would feed a subclinical dose [of molnupiravir] to people infected.”
Two other recent studies also called out molnupiravir, questioning its effectiveness and raising concerns the drug may help lead to the development of new COVID-19 variants.
A December 2022 preprint by a team of Australian researchers, found “this commonly used antiviral can ‘supercharge’ viral evolution in immunocompromised patients, potentially generating new variants and prolonging the pandemic.”
And a study published Jan. 28 in The Lancet found, “Molnupiravir did not reduce the frequency of COVID-19-associated hospitalisations or death among high-risk vaccinated adults in the community.”
University of Cambridge clinical microbiologist Ravindra Gupta, Ph.D., told Science that while it’s unclear whether molnupiravir will cause deadlier COVID-19 variants, the overall results of these recent studies “call into question whether molnupiravir should be used.”
Merck spokesperson Robert Josephson defended the product, telling Bloomberg, “There is no evidence that any antiviral agent has contributed to the emergence of circulating variants.”
Molnupiravir ‘different’ than Paxlovid — and ‘riskier’
Although molnupiravir is similar to Paxlovid in that both are oral antiviral treatments for COVID-19, Hooker told The Defender there are significant differences in how the two drugs work:
“Molnupiravir acts on the SARS-CoV-2 virus by directly inducing mutations in the RNA genome. This is a completely different mode of action compared to Pfizer’s product, Paxlovid, and in my estimation is quite dangerous.
“Merck claimed the mutation rate induced by molnupiravir would kill the virus and that mutants wouldn’t escape, but that has been shown to be false in studies of immunocompromised patients.”
Hooker said Paxlovid — and the COVID-19 vaccines — can potentially lead to the development of mutations as well.
But in his view, the “mechanism of action” used by molnupiravir is different — and far riskier — than Paxlovid and COVID-19 vaccines, which merely increase the virus’ lifetime in the human body, giving the virus a greater opportunity to naturally mutate.
Hooker said:
“In contrast, molnupiravir directly induces mutations and thereby vastly increases the mutation rate of the virus in the human host.
“In my estimation, this is a very dangerous way to treat such an infection, given the implications of creating random mutants.”
Merck made billions from molnupiravir — thanks to taxpayers
In 2022, sales of Merck’s molnupiravir hit $5.68 billion, fueled in part by strong fourth-quarter sales of the drug in Asia.
Fourth-quarter sales of molnupiravir reached $825 million, more than doubling analyst expectations of $358 million.
These strong earnings were boosted by government — or taxpayer — support.
In June 2021 — with molnupiravir still in clinical trials, which weren’t completed until October 2021 — the federal government signed a $1.2 billion contract with Merck for 1.7 million courses of the drug, at a cost of approximately $712 per patient.
An analysis by Melissa Barber of the Harvard T.H. Chan School of Public Health and Dzintars Gotham of King’s College Hospital in London found the cost of production of molnupiravir was approximately $1.74 per unit — or $17.74 for a five-day regimen.
By those calculations, the U.S. government paid a near-4,000% markup.
In March 2022, during his State of the Union address, President Biden announced the “Test to Treat” initiative, which allowed those who tested positive for COVID-19 at a pharmacy to obtain free antiviral pills — including molnupiravir — on the spot.
One month earlier, the Biden administration had proceeded with a new purchase of 3.1 million courses of molnupiravir, with the option to purchase more.
Estimates for Merck, and other COVID-19 drugmakers, are less rosy for 2023, as the public tires of all things pandemic and Biden looks to end the COVID-19 national emergency in May.
According to Reuters, sales of molnupiravir are expected to fall to about $1 billion this year, contributing to an expected decline in sales for Merck from $59.3 billion in 2022 to $57.2-$58.7 billion this year.
Merck’s stock price dropped by about 2% with Thursday’s announcement.
Despite these large earnings, overall sales of molnupiravir lagged significantly behind Paxlovid in 2022. Sales of Paxlovid reached $18.9 billion last year.
Michael Nevradakis, Ph.D., based in Athens, Greece, is a senior reporter for The Defender and part of the rotation of hosts for CHD.TV’s “Good Morning CHD.”
This article was originally published by The Defender — Children’s Health Defense’s News & Views Website under Creative Commons license CC BY-NC-ND 4.0. Please consider subscribing to The Defender or donating to Children’s Health Defense.
The Censorship of Mercola — A Timeline
By Dr. Joseph Mercola | January 30, 2023
While the drug and chemical industries have attacked and tried to discredit me for years, blatant censorship didn’t begin until 2020, after the outbreak of the COVID pandemic.
For legal and historical purposes, I am sharing a timeline of events with you that document a chain of coordinated events and attacks against me and this website. My first article about the pandemic came out February 4, 2020, in which I predicted that it was a grossly exaggerated threat that would enrich pandemic vaccine makers.
March 8, 2020, I published an interview with bioweapons expert Francis Boyle, Ph.D., in which he warned that SARS-CoV-2 had all the hallmarks of a genetically engineered bioweapon. Boyle was among the first, if not the first, to suspect the outbreak was the result of a lab leak.
While every health authority on the planet insisted there was no treatment, and that patients simply go to the hospital to be placed on mechanical ventilation and die, I interviewed medical experts working on early treatment options and published articles detailing the potential benefits of vitamin D, zinc, quercetin and other nutraceuticals that boost immune function, as well as decades-old drugs like hydroxychloroquine.
I also published the testimony of whistleblowers such as Erin Marie Olszewski, a frontline nurse, who warned that patients were being intentionally killed on ventilators as it quickly proved to be a deadly intervention for COVID-19.
Summer of 2020 — The Suppression of Vitamin D Begins
Early on, it became apparent that vitamin D levels and spending time outdoors played an important role in the risk of infection and the ultimate outcomes. This has been true for all respiratory infections, so it should come as no surprise it is also true for coronavirus infections. Despite that, health authorities insisted vitamin D was useless.
The only way out of the pandemic, they said, would be a vaccine — and this despite the fact that no previous attempts at creating a safe and effective coronavirus vaccine had ever succeeded because of its rapid ability to mutate.
In June 2020, I launched an information campaign, StopCovidCold, about vitamin D. I released a downloadable scientific report detailing how and why optimizing vitamin D levels among the general population could minimize the impact of the next wave of COVID. Optimizing vitamin D is a rational, safe and inexpensive measure that no sane health official would object to. And yet, they all did.
July 21, 2020, the Center for Science in the Public Interest (CSPI) issued a press release1 calling on the U.S. Food and Drug Administration and the Federal Trade Commission (FTC) “to bring enforcement proceedings against Mercola and his companies for their unlawful disease claims that falsely and misleadingly claim to treat, cure or prevent COVID-19 infections.”
CSPI accused me of falsely claiming “that at least 22 vitamins, supplements and other products available for sale on his web site can prevent, treat, or cure COVID-19 infection.” This despite the fact that their Appendix of Illegal Claims2 clearly show I made no COVID-19-related claims to any specific products and only referenced published studies and mainstream media articles to support my opinions.
In an August 12, 2020, email, CSPI president Dr. Peter Lurie — a former FDA associate commissioner — also made the spurious claim that I was “profiting from the pandemic” through “anti-vaccine fearmongering:” 3
“Mercola brazenly has claimed that many of his products are coronavirus treatments or cures, including vitamin C, vitamin D, zinc, selenium, ‘molecular hydrogen,’ licorice, and other substances.
Besides profiting from the pandemic, Mercola has seemingly advised people to contract COVID-19 after taking supposedly ‘immunity boosting’ supplements (which of course he sells). Making matters worse, Mercola is a leading proponent of anti-vaccine conspiracy theories — and has been fearmongering against prospective COVID-19 vaccines even before such vaccines are available!”
By mid-August, a comprehensive campaign to put an end to Mercola.com had been launched, with Laurie asking CSPI members to flood the FDA and FTC with prewritten Tweets, urging them to take action against us. He also urged “state attorneys general to investigate how they may further protect consumers from Mercola’s illegal marketing.”4
Not surprisingly, CSPI is funded by the Rockefeller Foundation, the Rockefeller Family Fund, Bloomberg Philanthropies and other billionaire-owned foundations. It’s also partnered with Bill Gates’ agrichemical PR group, the Cornell Alliance for Science. Greg Jaffe, who heads up CSPI’s Biotechnology Project, is also the associate director of legal affairs at Alliance for Science.
Winter of 2020 — Vitamin D Attacks Heat Up
The attacks against me really heated up though after I published a peer-reviewed scientific paper5 on the benefits of vitamin D at the end of October 2020. With that, I established my medical and scientific merit and my right to a professional opinion, which is something the U.S. Constitution absolutely provides for.
The paper, “Evidence Regarding Vitamin D and Risk of COVID-19 and Its Severity,” published in the journal Nutrients, was coauthored by William Grant, Ph.D., and Dr. Carol Wagner, both of whom are part of the GrassrootsHealth expert vitamin D panel.
As noted in that paper, dark skin color, increased age, pre-existing chronic conditions and vitamin D deficiency are all features of severe COVID disease, and of these, vitamin D deficiency is the only factor that is modifiable. As such, it would be foolish to ignore, especially since vitamin D supplements are readily available and low cost.
Christmas Eve 2020, attorney general Letitia James sent us a cease and desist notice, demanding we stop sharing information about how immune-boosting nutritional supplements might lower your risk of COVID, including vitamin D, zinc, NAC and vitamin C.
February 2021, FDA Tries to Silence Protected Speech
After the new presidential administration took over, on February 18, 2021, the Rockefeller-funded CSPI and AG James got their wish, as the FDA sent us a warning letter for “Unapproved and Misbranded Products Related to Coronavirus Disease 2019.”6 Laurie even publicly bragged7 about his ability to influence the FDA to take action against us.
The FDA’s letter highlighted statements in articles on my website that were fully referenced and supported by published science, and none of the articles cited had any commercial advertising linking the information to my products, as per the law. We had done nothing illegal or irregular in that regard, and my professional opinions are protected under the U.S. Constitution.
Needless to say, we fully addressed both James’ cease and desist notice and the FDA’s warning letter, putting them both on notice that they cannot censor protected speech simply because they don’t like what’s being said.
On a side note, William Correll, the director of the Office of Compliance at the FDA who signed the warning letter, sadly “passed away suddenly” just two months later, on April 18 “after a short battle with COVID-19.”8
Gates-Funded Front Group Gets on the Bandwagon
The agrochemical front group Cornell Alliance for Science (CAS),9 the primary funding for which comes from the Bill & Melinda Gates Foundation,10 also jumped on the bandwagon, falsely stating11 that “pages advertising vitamin C and quercetin as having ‘synergistic effects that make them useful in the prevention and early at-home treatment of COVID-19′” were still available on my website nearly a month after the FDA’s warning letter.
To be clear, we had fully referenced scientific news articles. News articles are NOT “advertising,” as they do not link to any specific products, nor do they refer to or recommend any specific brands. In the case of the warning for vitamin C, the article discussed hospitals utilizing IV vitamin C for the treatment of COVID-19 and sepsis.
Such coordinated attacks are to be expected, though, considering Gates’ influence over the operation, and seeing how CAS and CSPI work closely together — a fact CAS admitted in its hit piece.12
March 2021, Booksellers Urged to Ban My Book
Around that same time (February 11, 2021), my book “The Truth About COVID-19” also went up for presale, and by early March, booksellers in the U.S., U.K. and Australia were being pressured not to sell it, or to add some sort of misinformation warning label to it. As reported by Sky News March 5, 2021:13
“In the UK, more than 20 million vaccine doses have been administered as part of efforts to defeat COVID-19, but worries continue that misinformation is stopping some people from having the jab. Shadow health minister Alex Norris told Sky News:
‘Getting our population vaccinated is a massive priority and it is very sad to see these things so freely available. We would hope that retailers would act responsibly and have a look at whether they want to be associated with such products and whether they want to be seen to be profiting off such products.'”
Shady ‘Anti-Hate’ Outfit Publishes Hit List
March 3, 2021, the Center for Countering Digital Hate (CCDH) — a shady U.K.-based organization with anonymous funding led by Imran Ahmed — also got in on the action, publishing a hit list14 of the “Top 10 anti-vaxxers” it wanted permanently silenced and eradicated from public forums. The list showed, by way of crossing out names, which had already been successfully deplatformed, and from which social media.

While precious little was (and still is) know about the CCDH, some digging revealed Ahmed had been appointed to the steering committee of the U.K. government’s Commission on Countering Extremism Pilot Task Force in April 2020, just as fearmongering about the COVID-19 pandemic was ramping up. The CCDH is also linked to a number of technocratic centers within the globalist network through its board members.15
More Fabrications and Lies From the CCDH
A couple of weeks later (March 15), Ahmed somehow managed to get an article titled “Dismantling the Anti-Vaxx Industry”16 published in the journal Nature Medicine. In it, Ahmed lied, claiming he’d “recorded a private, three-day meeting of the world’s most prominent anti-vaxxers,” when in fact it was a public, international conference given online, attended by thousands around the world, all of whom had access to the recordings.
He could have done the normal, ethical and truly journalistic thing and admitted he simply attended a public virtual conference, but instead he twisted it into some risky undercover agent mission where he secretly recorded private discussions that revealed the inner workings of “the opposition.”
Then, March 21, 2021, the CCDH published the fabricated “Disinformation Dozen” report,17,18,19 in which Ahmed falsely claimed 12 people and/or organizations, including yours truly, were responsible for 65% of all anti-vaccine content on social media.
March 24, 2021 — AGs Try to Censor Protected Speech
March 24, 2021, 12 attorneys general sent a letter20 to the CEOs of Twitter and Facebook, seeking their “cooperation in curtailing the dissemination” of COVID jab “misinformation” — all based on the fabrications of the CCDH. According to the AGs:
“The people and groups spreading falsehoods and misleading Americans about the safety of coronavirus vaccines are threatening the health of our communities, slowing progress in getting our residents protected from the virus, and undermining economic recovery in our states.
As safe and effective vaccines become available, the end of this pandemic is in sight. This end, however, depends on the widespread acceptance of these vaccines as safe and effective. Unfortunately, misinformation disseminated via your platforms has increased vaccine hesitancy …
According to a recent report by the Center for Countering Digital Hate, so-called ‘anti-vaxxer’ accounts on Facebook, YouTube, Instagram and Twitter reach more than 59 million followers … Given ‘anti-vaxxers’ reliance on your platforms, you are uniquely positioned to prevent the spread of misinformation about coronavirus vaccines …”
Facebook Set the Record Straight
August 18, 2021, after conducting an internal investigation, Monika Bickert, vice president of Facebook content policy, publicly called out the falsehoods in “The Disinformation Dozen” report, stating:21
“In recent weeks, there has been a debate about whether the global problem of COVID-19 vaccine misinformation can be solved simply by removing 12 people from social media platforms. People who have advanced this narrative contend that these 12 people are responsible for 73% of online vaccine misinformation on Facebook.
There isn’t any evidence to support this claim … In fact, these 12 people are responsible for about just 0.05% of all views of vaccine-related content on Facebook. This includes all vaccine-related posts they’ve shared, whether true or false, as well as URLs associated with these people.”
Bickert highlighted the fact that Ahmed had preselected the 12 individuals listed in the report, and that his “faulty narrative” was based on nothing more than “a narrow set of 483 pieces of content over six weeks from only 30 groups, some of which are as small as 2,500 users.”
“Further, there is no explanation for how the organization behind the report identified the content they describe as ‘anti-vax’ or how they chose the 30 groups they included in their analysis,” Bickert noted. “There is no justification for their claim that their data constitute a ‘representative sample’ of the content shared across our apps.”
Apparently, no one in government was smart enough to see the flaws in the CCDH’s report though, and a long list of officials cited the CCDH’s fabricated claims throughout the remainder of 2021, even long after Facebook denounced its claims. What’s more, even though Facebook admitted the CCDH’s claims were bogus, they still took action against accounts by applying penalties and/or bans.
April 8, 2021 — AGs Call on Social Media to Ban ‘the 12’
April 8, 2021, attorneys general James and William Tong published an op-ed in The Washington Post,22 again calling on social media companies to ban the “disinformation dozen” identified by the CCDH. The lack of acceptance of novel gene therapy technology, they claimed, was all because a small group of individuals with a social media presence — myself included — were successfully misleading the public with lies about nonexistent vaccine risks.
April 27, 2021 — Dr. Hotez Calls for Cyberwarfare
April 27, 2021, Dr. Peter Hotez, president of the Sabin Vaccine Institute23 — which has received tens of millions of dollars from the Bill & Melinda Gates Foundation,24,25 — escalated the threat even further in an article published in the journal Nature.
Citing the CCDH’s findings, Hotez called for cyberwarfare experts to be enlisted in the war against vaccine safety advocates and people who are “vaccine hesitant.” He wrote:26
“Accurate, targeted counter-messaging from the global health community is important but insufficient, as is public pressure on social-media companies. The United Nations and the highest levels of government must … move to dismantle anti-vaccine groups in the United States.
Efforts must expand into the realm of cyber security, law enforcement, public education and international relations. A high-level inter-agency task force reporting to the UN secretary-general could assess the full impact of anti-vaccine aggression, and propose tough, balanced measures.
The task force should include experts who have tackled complex global threats such as terrorism, cyber attacks and nuclear armament, because anti-science is now approaching similar levels of peril. It is becoming increasingly clear that advancing immunization requires a counteroffensive.”
In short, Hotez called for the use of warfare tactics on law abiding American citizens, and the Nature journal actually published this blatant threat. One day later, April 28, the CCDH published a second report, “Disinformation Dozen: The Sequel,”27 which focused on Big Tech’s failure to get rid of us “despite bipartisan calls from Congress.”
To understand the massive reach the CCDH gained, despite no one having heard of them before COVID, consider this: By the end of August 2021, there were 84,700 Google search results for CCDH’s defamatory phrase “disinformation dozen,” including 16,000 news stories in the international press, nearly all of which parroted the CCDH’s defamatory statements verbatim and reported them as fact.
May 2021 — Financial Warfare Led to Removal of COVID Articles
Shortly after the op-ed by AGs James and Tong appeared, our business bank accounts were abruptly shut down and our credit cards canceled. Our business partners also had their PayPal accounts shut down.
This new threat, which I could not defend against in a court of law, led to my May 4 decision to remove all articles related to vitamin D, vitamin C, zinc and COVID-19 from my website.
July 2021 — The White House Publicly Calls for Censorship
In mid-July 2021, the White House stepped in to pressure Facebook to purge “anti-vaxxers” from its platform. Then-press secretary Jen Psaki regurgitated the CCDH’s false claims, saying:28
“There’s about 12 people who are producing 65% of anti-vaccine misinformation on social media platforms. All of them remain active on Facebook, despite some even being banned on other platforms, including ones that Facebook owns.
Facebook needs to move more quickly to remove harmful, violative posts. Posts that would be within their policy for removal often remain up for days, and that’s too long. The information spreads too quickly.”
In another mid-July press conference, President Joe Biden himself demanded social media take action against “the disinformation dozen,” claiming our “misinformation” was “killing people.”29,30 None of these officials ever questioned the authority of the CCDH. Facebook spokesperson Dani Lever responded to the White House’s demands, saying:
“We will not be distracted by accusations which aren’t supported by the facts. The fact is that more than 2 billion people have viewed authoritative information about COVID-19 and vaccines on Facebook, which is more than any other place on the internet … The facts show that Facebook is helping save lives. Period.”
Summer of 2021 — A Parade of Hit Pieces
July 24, 2021, the New York Times named me the No. 1 superspreader of COVID misinformation online.31 According to the NYT itself, this was the most-read article of the year up to that point. Penned by Sheera Frenkel, it was so littered with blatant lies, my attorneys sent her a retraction demand.32
For example, she claimed the FDA has levied multimillion-dollar fines against me. This is a complete fabrication, as I’ve never been fined by the FDA. She also implied that I misrepresented myself as a published author of a paper on vitamin D for COVID-19, stating she was “unable to verify” my claim. This despite being given a direct link to the paper! My paper can also be located on PubMed.gov in seconds by searching my name.
Frenkel boldly claimed that I am the No.1 spreader of misinformation online, but she didn’t even qualify what “misinformation” actually is. Without qualifying what it is you’re looking for, how can you quantify it? She also provided no proof that I in fact had the greatest reach of all the individuals reporting on COVID injections. My name didn’t even show up in the Top 15 in a Crowdtangle search for anti-vax Facebook posts.
Frenkel’s hit piece was followed up by CNN, which August 4 aired a segment show CNN reporter Randi Kaye stalking me across central Florida. And, of course, Kaye’s primary citation for her accusations against me was the CCDH.
August 4, 2021 — Mercola Deletes Articles After 48 Hours
August 4, 2021, I also implemented yet another change on my website. I had already removed all articles relating to COVID-19 and vitamin D. At this point, I deleted over 15,000 articles from the past 20-plus years from my website as the business and personal threats grew out of hand.
After 48 hours, articles were instead migrated over to Substack, where only paid members through a private membership agreement have access to them. This was a painful but necessary workaround, as the paid subscription provides a layer of protection against these threats.
September 7, 2021 — Senator Warren’s Book Burning Campaign
September 7, 2021, U.S. Sen. Elizabeth Warren sent a letter33 to Andy Jassy, chief executive officer of Amazon.com, demanding an “immediate review” of Amazon’s algorithms to weed out books peddling “COVID misinformation.”34,35,36
While she didn’t spell out what laws Amazon might be breaking, she warned Jassy that the company may be held legally responsible for wrongful death and homicide by selling books that “misinform” readers about COVID-19, and she specifically singled out “The Truth About COVID-19” as a prime example of the kinds of books she wanted banned.
Warren again relied on the fabrications of the CCDH, even though Facebook had refuted the CCDH report as baseless three weeks before she sent that letter.
“Dr. Mercola has been described as ‘the most influential spreader of coronavirus misinformation online,'” Warren wrote, adding: “Not only was this book the top result when searching either ‘COVID-19’ or ‘vaccine’ in the categories of ‘All Departments’ and ‘Books’; it was tagged as a ‘Best Seller’ by Amazon and the ‘#1 Best Seller’ in the ‘Political Freedom’ category.
The book perpetuates dangerous conspiracies about COVID-19 and false and misleading information about vaccines. It asserts that vitamin C, vitamin D and quercetin … can prevent COVID-19 infection … And the book contends that vaccines cannot be trusted, when study after study has demonstrated the overwhelming effectiveness and safety of COVID-19 vaccines.
It should come as no surprise that the book is rife with misinformation. One of the authors, Dr. Mercola, is one of the ‘Disinformation Dozen,’ a group responsible for 65% of anti-vaccine content on Facebook and Twitter …”
YouTube Deplatforms Mercola in Breach of Contract
Warren’s attempt at getting Amazon to ban my book was swiftly followed up by YouTube, which deleted my account September 29, 2021, allegedly for violating community guidelines. The problem was, they’d published and implemented those new guidelines that very morning.
While I disagreed with YouTube’s censorship, when its “COVID-19 misinformation” policy was implemented back in April 2021, I carefully avoided posting any content on YouTube that might violate that guideline. At no point had I ever received a violation notice from YouTube.
On the morning of September 29, 2022, at 9 a.m. EDT, The Washington Post published an article titled “YouTube Is Banning Joseph Mercola and a Handful of Other Anti-Vaccine Activists.” According to the WaPo :37
“YouTube is taking down several video channels associated with high-profile anti-vaccine activists including Joseph Mercola … As part of a new set of policies aimed at cutting down on anti-vaccine content on the Google-owned site, YouTube will ban any videos that claim that commonly used vaccines approved by health authorities are ineffective or dangerous.
The company previously blocked videos that made those claims about coronavirus vaccines, but not ones for other vaccines like those for measles or chickenpox.”
In short, as of September 29, 2021, you could no longer post any video discussing or stating that any vaccine is dangerous or ineffective. Six minutes after the publication of that WaPo article, I received an email from YouTube informing me that my entire channel had been deplatformed, having been found in violation of this new policy.
October 2021 — CNN’s Second Hit Piece
October 4, 2021, two months to the day after their first attempted hit piece against my book, “The Truth About COVID-19,” CNN aired a follow-up in which they echoed Warren’s call for Amazon to ban the sale of my book.
Like something straight out of George Orwell’s “1984” newsspeak dictionary, CNN host Anderson Cooper said my book is loaded with “mistruths” about COVID. Yet he failed to present a single piece of evidence to back up that claim.
This is one of the oldest propaganda trick in the book. If you just spew out enough derogatory terms about your opponent, people will forget the fact that you provided zero proof to back up your position.
November 2021 — Mercola Sues Sen. Warren
November 7, 2021, two months after Warren tried to get my best-selling book “The Truth About COVID-19” banned from Amazon, I, my coauthor Ronnie Cummins, my publisher and Robert F. Kennedy Jr., who wrote our foreword, sued Warren,38 both in her official and personal capacities, for violating our First Amendment rights and scaring book sellers into pulling and/or suppressing sales.
As a government official, it is illegal for her violate the U.S. Constitution, and pressuring private businesses to do it for her is not a legal workaround.
February 2022 — NIH Director Blames Mercola For Pandemic Continuation
In February 2022, former National Institutes of Health director Dr. Francis Collins blamed me personally for the government’s inability to bring the COVID pandemic to a close. This despite the fact that I was by then heavily censored just about everywhere. The only people, really, who could see my information were those who subscribed to my newsletter and received it by email.
August 2022 — NYT Airs Hit Piece Documentary
Fast-forward to August 2022, The New York Times published the documentary “Superspreader,” featuring yours truly, on FX and Hulu (both of which are owned by Disney). They clearly went through a lot of trouble, trying to dig up dirt from anyone they could find from my past — some going back 40 years, to my medical school days — who would be able to share some tidbit with which they could discredit me with.
But it seems they came up empty handed: After a year of investigation, they couldn’t come up with anything. Surprisingly, they even showed two people who claimed I’d saved their lives. All the other interviews were with people who don’t actually know me. One was with a Chicago journalist who interviewed me once — 13 years ago. Two classmates from med school, whom I haven’t seen in over 40 years, also described their impressions.
Ironically, yet again, just one week before the “Superspreader” program aired, the U.S. Centers for Disease Control and Prevention reversed all of its COVID-19 guidelines, thereby proving my position on COVID was correct all along. Of course, this was never mentioned in their program though.
September 23, 2022 — Mercola Website Taken Down in Cyberattack
Next up was a cyberattack that took down my entire website and destroyed our servers. Cyberattacks have been ongoing for the past six years, but the one that took place September 23, 2022, finally got through our defenses. By that time, my reach on social media had been throttled back to next to nothing, and my website was about the only place you could find my articles (with the exception of republications, which I allowed).
September 28, 2022 — Mercola Sues Google and YouTube
Warren isn’t the only one I’ve had to sue to protect my First Amendment right. In September 28, 2022, I also filed a lawsuit39 against Google, YouTube and Alphabet Inc. for breach of contract.40
As detailed in my complaint, YouTube unilaterally amended the contract without notice, which is a violation of its own terms of service, and then used this last-minute amendment to justify removing my content, which went back to 2005, the same year YouTube was founded. At the time YouTube deleted my content, I had more than 300,000 subscribers, and my videos had collectively garnered more than 50 million views.
The WaPo article was embargoed until the morning of September 29 in order to prevent me (and anyone else affected by this change) from reviewing the new policy, take steps to bring my channel into compliance, or move my content to another platform. Instead, they simply deleted 16 years’ worth of intellectual property, without warning.
This is a clear violation of its own terms of service, which state that YouTube “will provide reasonable advance notice” of any changes to the terms of service, and that users will have “the opportunity to review them” and to remove content if they do not agree to the new terms.
YouTube’s terms of service also include a “three strikes” policy, where users are given three warnings and opportunities to remove content that violates the guidelines before being banned. I had no “strikes” against my channel on the day I was deplatformed and deleted.
I’m also suing YouTube for unjust enrichment, as for the last 16 years, my video content, having generated in excess of 50 million views, has been of great financial benefit to YouTube, allowing them to increase advertising revenue on the site. Additionally, they’ve refused to allow me to retrieve any of this content, which they still have in their possession. So, YouTube has unjustly benefited at my expense.
January 2023 — Third Lawsuit Filed to Protect Free Speech
January 10, 2023, I, along with several other plaintiffs, also filed a lawsuit41 against The Washington Post, the BBC, the Associated Press and Reuters — also known as the Trusted News Initiative (TNI),42 a self-appointed Pharma and Big Tech industry partner that has spent the past couple years playing judge and jury of news.
It has been doing everything it can to censor what it doesn’t want the public to hear. As noted in the complaint, the TNI has not only censored free speech, it has also engaged in antitrust activity. Specifically, “Federal antitrust law has its own name for this kind of ‘industry partnership’: it’s called a ‘group boycott’ and is a per se violation of the Sherman Act.”
As evidence of this allegation, our complaint references multiple public statements by TNI partners, including a March 2022 statement by Jamie Angus, then-senior news controller for BBC News, who explained TNI’s “strategy to beat disinformation.”
The Fight for Truth and Freedom Continues
The globalist cabal is extremely coordinated, as you can see. What’s more, they play dirty. But we will not give up, nor give in. Our freedom is far too precious for that, and freedom depends on getting the truth out. So, I will continue doing my part. You can help by sharing articles you think are important with family and friends, in whatever ways are available.
Sources and References
- 1 CSPI July 21, 2020
- 2 Illegal Claims Pertaining to Mercola Group Products (PDF)
- 3 CSPI July 19, 2017
- 4, 7 SCPInet.org March 4, 2021
- 5 Nutrients October 31, 2020;12, 3361; doi:10.3390/nu12113361
- 6 FDA Mercola Warning Letter February 18, 2021
- 8 Sun Gazette April 24, 2021
- 9 USRTK September 23, 2020
- 10 Cornell Alliance for Science Our Funders
- 11, 12 Cornell Alliance for Science, March 2021, FDA Warns Mercola
- 13 Sky News March 5, 2021
- 14 Twitter CCDH March 3, 2021
- 15 In-this-together.com, CCDH The Center for Cancel Culture and Digital Hypocrisy Part 2
- 16 Nature Medicine March 15, 2021
- 17 Counterhate.com Disinformation Dozen March 21, 2021
- 18 The Disinformation Dozen
- 19 PR Newswwire March 24, 2021
- 20 Letter to Jack Dorsey and Mark Zuckerberg March 24, 2021
- 21 Facebook August 18, 2021
- 22 Washington Post April 8, 2021
- 23 Texas Children’s Hospital Peter Hotez
- 24 PND July 1, 2011
- 25 Bill & Melinda Gates Foundation
- 26 Nature April 27, 2021
- 27 CCHD Disinformation Dozen: The Sequel April 28, 2021
- 28 Reason July 15, 2021
- 29 CNN July 16, 2021
- 30 AP July 16, 2021
- 31 New York Times July 24, 2021 (Archived)
- 32 Twitter Dr. Joseph Mercola July 26, 2021
- 33 Warren’s letter to Andy Jassy September 7, 2021
- 34 National Interest September 12, 2021
- 35 The Guardian September 13, 2021
- 36 New York Times September 8, 2021
- 37 Washington Post September 29, 2021
- 38 US District Court Western District of Washington Case: 2:21-cv-01508
- 39 Complaint for Contract Breach Mercola vs Google, YouTube, Alphabet Inc. September 28, 2022
- 40 Reuters September 29, 2022
- 41 US District Court Northern District of Texas Case: 2:23-cv-00004-Z
- 42 The Defender January 10, 2023
FDA commissioners say the agency needs ways to fight online “misinformation”
By Cindy Harper | Reclaim The Net | January 10, 2023
Former and current Food and Drug Administration (FDA) commissioners said that the agency needs partners to fight public health misinformation and that patient advocates, clinicians, industry, and academic leaders have a role to play.
The commissioners made the comments at the 2023 Innovations in Regulatory Science Summit, an event that was organized by the UCSF-Stanford Center of Excellence in Regulatory Science and Innovation (CERSI).
“I actually believe that misinformation is the leading cause of death right now in the US because whether we’re looking at COVID or chronic disease, people are making bad choices driven by the information that they get,” said FDA Commissioner Robert Califf, as reported by regulatory focus. “We were just not prepared for what broad access to the internet would do to communication channels.”
Califf said that the academic community was not doing enough to combat misinformation and that their criticism of the FDA is having unintended consequences.
“As a public agency, we need to be critiqued but I think often the people that are doing the critiquing assume that the agency’s going to be there in the future in the way that they expect it to be there,” Califf said. “So, they’re critiquing it to make it better. But to a lot of unsuspecting people that hear it, it just completely erodes their belief in the institution.”
Mark McClellan, who served as an FDA commissioner from 2002 to 2004 said, “Realistically, FDA needs help.” He acknowledged that there is currently a lack of trust in public health agencies and officials. However, people still trust their doctors, community leaders, and others that are “close to their experience.”
Scott Gottlieb, a Pfizer board member who served as a commissioner from 2017 to 2019, said the fast response to misinformation is crucial and touted the idea of allowing the industry to counter misinformation about products.
“We’ve seen FDA weigh in, admirably, around some dangerous disinformation on specific products,” he said. “But that can’t be the business of the FDA.”
He suggested that the FDA should create a limited safe harbor to allow sponsors to directly counter misinformation. He added that the FDA would determine how and what the sponsors can respond to.
“I think sponsors need to have the ability to defend their products in the marketplace of ideas when there’s true misinformation,” Gottlieb said.
Gottlieb was under fire this week after it was revealed that Gottlieb had been flagging tweets to Twitter.
In the August 27, 2021 email, which was published by journalist Alex Berenson, Gottlieb complained to Todd O’Boyle, a senior manager on Twitter’s Public Policy team, about a tweet that claimed natural immunity to Covid-19 was superior to vaccine immunity.
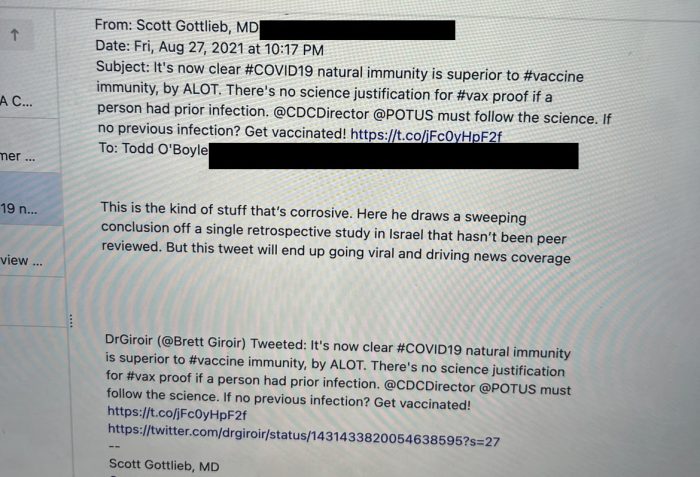
“This is the kind of stuff that’s corrosive,” Gottlieb wrote. “Here he draws a sweeping conclusion off a single retrospective study in Israel that hasn’t been peer reviewed. But this tweet will end up going viral and driving news coverage.”
Alzheimer’s drug approval raises the alarm
Data shows treatment can lead to ‘brain shrinkage’
By Maryanne Demasi, PhD | January 9, 2023
The US Food and Drug Administration (FDA) has granted an accelerated approval of a new treatment for Alzheimer’s disease, which aims to clear toxic amyloid protein build-up in the brain.
At a cost of $26,500 per year in the US (not covered by Medicare or Medicaid), people with early Alzheimer’s disease can receive a twice-monthly monoclonal antibody infusion called lecanemab (marketed as LEQEMBI™), co-developed by Eisai, a Japanese biotech firm, and Biogen.
In the lead up to the FDA’s approval, there was intense lobbying for the drug.
A ‘consensus statement’ signed by over 200 scientists, many of whom had financial ties to the drug companies, described lecanemab as a “foundational gamechanger” for the disease, calling for “no barrier” to the widespread availability of the treatment.
Now that the drug has been approved, advocacy groups like the Alzheimer’s Association, which are heavily funded by the drug industry, have welcomed the news, saying the FDA made “the right decision.”
But critics doubt the benefits of lecanemab outweigh its harms, and are dismayed that the FDA approved the drug without input from its own advisory panel.
Kim Witczak, a drug safety advocate, and member of the FDA’s Psychopharmacologic Drugs Advisory Committee, says she is “shocked” by the latest FDA stunt.
“By approving this new drug without a public advisory committee meeting, the FDA once again has shown a lack of concern for the public, patients, and healthcare providers. Convening its advisory panel would have helped reassure everyone that the FDA’s decision was scientifically sound and transparent,” said Witczak.
“Advisory committee meetings offer the opportunity to discuss the data in an open and public forum, to challenge methods, study endpoints (surrogate vs clinically meaningful), and safety findings before the general committee member discussion. But in this case, none of that was possible,” she added.
The FDA’s accelerated approval process used to green-light lecanemab is known for accepting lower evidentiary standards for drug efficacy, so that patients can gain access to experimental drugs sooner.
Critics say its reminiscent of the FDA’s approval of aducanumab – Biogen’s other Alzheimer’s drug. It was approved on the basis of lowering amyloid protein (a surrogate marker) in the brain, despite no clinically meaningful benefit for patients.
At the time, the controversial decision led to the resignation of FDA advisory member Aaron Kesselheim, who labelled it “probably the worst drug approval decision in recent U.S. history.”
Linda Furlini, a research ethics advisor based in Montreal, Canada says it essentially gives the rubber stamp to similar drugs down the track. “Once you grant accelerated approval of a drug in that class, then it’s easier to get the second drug, and then the third drug approved.”
Jessica Adams, an expert in drug regulatory affairs, agrees. She said, “Lecanemab’s approval shows the power of precedent in regulatory approvals. This is why I scoff whenever the FDA says it still reviews drugs on a case-by-case basis.”
The industry-funded study published in the New England Journal of Medicine, involving almost 1800 people with early Alzheimer’s disease, found that lecanemab could slow the decline of cognition and function by 27% over 18 months compared to placebo.
They used a “Clinical Dementia Rating” scale to show lecanemab patients declined by 1.21 points compared to 1.66 point in the placebo group – a 0.45 point difference in lecanemab’s favour.
But experts question whether the small difference will have any impact on how the patient actually feels.
Madhav Thambisetty, a neurologist at Johns Hopkins University and the National Institute on Aging said, “The benefit appears to be quite small, and it’s unclear how meaningful this might be for patients.”
In fact, the FDA’s own statistician Dr Tristan Massie was uncertain whether “the treatment effect on amyloid is reasonably likely to predict change on the clinical outcome” and considered the results of the study to be “exploratory”.
As a physician who cares for people with Alzheimer’s disease, Thambisetty spoke about the harms of the drug. “These patients can experience headaches, falls, confusion, vision disturbances and it’s unclear if patients will be able to see obvious benefits on a day-to-day basis,” he said.
The data showed an increased risk of brain bleeds and swelling, i.e. amyloid-related imaging abnormalities (ARIA) occurred in 126 (14.0%) of subjects in the lecanemab group and only 69 (7.7%) of subjects in the placebo group.
This prompted the FDA to include a warning on the drug about the risk of swelling and bleeding in the brain.
The drugmakers have also highlighted that people carrying two copies of the APOE4 gene (which predisposes someone to Alzheimer’s) puts them at a particularly “high risk of life-threatening brain haemorrhage.”
Three deaths have been reported in people taking lecanemab; an 80-yr old phase 3 trial participant who suffered intracranial haemorrhage, a 65-yr old who experienced brain swelling and bleeding and a 79-yr old who reportedly had seizures and brain bleed in the open-label phase of the trial.
Two of the three people who died were taking blood thinners, and experts who reviewed the lecanemab death cases suggested that anticoagulant use may have exacerbated the fatal outcomes.
Furlini’s research career has focused on the need to educate and support caregivers of people with dementia-type illnesses.
“You read the list of side effects – you might have gait problems, you might have brain swelling, visual disturbances… I mean, what are we doing here?” asks Furlini, “The patient is already confused and losing their cognitive capacity. How are these serious side effects helping them? It runs counter to any ethical semblance of what is wanted or expected.”
Thambisetty has also expressed concerns about the “brain shrinkage” seen in trial participants taking either lecanemab or aducanumab – increasing doses of the drug correlate to a decrease in brain volume.
“The observation of brain shrinkage is worrisome because, in the absence of compelling evidence to the contrary, it suggests a potential worsening of degenerative changes in the brains of people with Alzheimer’s disease,” wrote Thambisetty in a recent opinion piece for STAT.
The observation has been explained away by researchers who say that a reduced brain volume is due to the clearance of amyloid protein from the brain. But Thambisetty says there is little empirical evidence to support this theory.
Instead, he points to an Australian study which calculated that the clearance of amyloid plaque from the brain was too small to represent a plausible explanation for the loss of brain volume.
US lawmakers launched an investigation into the FDA after the agency’s controversial approval of aducanumab. Last month, a US House of Representatives panel released the report following an 18-month investigation.
The report said the process was “rife with irregularities” and that FDA officials “inappropriately collaborated” with the drugmaker during the approval process which “exceeded the norm in some respects.”
Representatives from the FDA and Biogen engaged in over 100 phone calls or meetings dating back to 2019 in order to expedite the drug’s approval, which lawmakers say, “consisted of atypical procedures and deviated from the agency’s own guidance.”
The congressional report recommended the agency “must take swift action to ensure that its processes for reviewing future Alzheimer’s disease treatments do not lead to the same doubts about the integrity of FDA’s review.”
But critics now say, it’s too late for an agency that has not taken accountability for its actions.
“These drug approvals have just created confusion, uncertainty, fear and misinformation. Then they wonder why people have no trust in their institutions, like the FDA. The world looks to the FDA for leadership. That it does not fulfill its responsibilities, remains the challenge of our times,” said Furlini.
Furlini has followed this area of research for decades and says the drug industry needs to move on from the ‘amyloid theory’ of Alzheimer’s disease and refocus its attention on other causes.
“After so many years, I’m fed up with the exclusive focus on the amyloid theory to the exclusion of other research theories, it’s a disservice to people with Alzheimer’s, and their families,” said Furlini
“There are a lot of buzzwords and marketing propaganda being put out there. And they justify it by saying that you have to give people hope. But you’re giving people false hope. It plays with people’s emotions, which I find horrendous,” added Furlini.
FDA Study: Pfizer Vaccine increases risk of Blood Clots in the lungs by 54% in the over 65s
The Naked Emperor’s Newsletter | January 9, 2023
The FDA recently published a study which looked at the COVID-19 vaccine safety among elderly persons aged 65 years and older. Today it was published in the Vaccine Journal.
The study looked at 30,712,101 individuals who had received various doses of the Pfizer, Moderna and Johnson & Johnson vaccines between 11 December 2020 and 15 January 2022.
Worryingly they identified four statistical signals for elevated risk of acute myocardial infarction (ACI), pulmonary embolism (PE), disseminated intravascular coagulation (DIC) and immune thrombocytopenia following the Pfizer vaccine. They didn’t find any statistical signals for the Moderna or Johnson & Johnson vaccines for the 14 outcomes they were monitoring.
PEs (blood clots on the lungs) were 54% more likely, ACIs (heart attacks) were 42% more likely, DICs (blood clotting disorder) were 91% more likely and ITPs (platelet disorder) 44% more likely.
However, they say that after further evaluation the rate ratios for AMI, DIC and ITP no longer met the statistical threshold for a signal. But, even after their further evaluation, the rate ratio for PE still met the statistical threshold.
Comically/Tragically, after more than two years of injecting people, they call their monitoring study an “early warning safety system”!
They conclude that this FDA “early warning safety system is working to rapidly identify potential new and important safety concerns following COVID-19 vaccination”. Wow, I’m glad they didn’t use their slower system because then we’d be in real trouble!
As usual and as you would expect, they go to great lengths to say that the four outcomes aren’t necessarily caused by the vaccine and may be related to other factors.
Furthermore, even though their own study has just shown the increased risks to the elderly, they say they BELIEVE the potential benefits of the vaccines outweigh the potential risks of Covid infection. Since when has ‘believe’ been a scientific way of analysing things? It sounds more like a religious conviction.
As a result, they won’t be taking any regulatory actions based on these signals, because they are still under investigation and require more robust study. So in the meantime, keep taking your boosters and we’ll let you know in maybe another two years that yes the blood clots in your lungs were from the vaccine. But we still BELIEVE your blood clots were better than your Covid infection, which you still got five times anyway – Amen.
I wonder which MSM outlets will report on the FDAs own study?
US regulator fast-tracks dementia drug
RT | January 6, 2023
The US Food and Drug Administration (FDA) on Friday fast-tracked the approval of lecanemab, a drug to treat the early stages of Alzheimer’s disease. Made by Japanese drugmaker Eisai and Biogen and marketed as Leqembi, the drug allegedly delays cognitive decline caused by the disease, though trials have shown some alarming side effects.
While a clinical trial of lecanemab’s efficacy in early Alzheimer’s published in November found it slowed cognitive and functional decline better than a placebo, the researchers noted that it was “associated with adverse events” and recommended “longer trials” to “determine the efficacy and safety of lecanemab in early Alzheimer’s disease” – an unusual call for caution in a study co-funded by the drug’s manufacturers.
Around 17% of those who took lecanemab experienced brain bleeding during the trials, while nearly 13% suffered brain swelling or effusions, compared to 9% and 2% in the placebo group respectively, according to the New England Journal of Medicine study. Some 7% of the trial participants stopped taking the drug due to the side effects.
Lecanemab’s high price point – $26,500 for a year’s worth of treatment – has also raised concerns. The Institute for Clinical and Economic Review suggested $20,600 as the price ceiling, arguing a cost-effective rate could be as low as $8,500. The company suggested it could lower the dosing frequency to cut costs.
Biogen is no stranger to controversy over its Alzheimer’s drugs. In 2021, several FDA board members resigned over concerns that Aduhelm, which the company had developed as the first drug designed to target the plaque buildup then believed to be the underlying cause of Alzheimer’s, had not demonstrated sufficient efficacy in treating moderate-to-severe dementia. While not a single member of the advisory panel responsible for reviewing the drug supported its approval, the FDA did so anyway, side effects and $56,000 annual price tag notwithstanding.
A congressional investigation that concluded last week found the approval process “rife with irregularities,” noting the FDA had “inappropriately collaborated” with the company it was supposed to be regulating.
Last year, it emerged that parts of the research that established the current plaque-based disease model of Alzheimer’s were possibly fraudulent, suggesting that the amyloid plaques found in the patients could be a symptom, rather than the cause, of the illness.
How to hide adverse events
FDA compared apples and oranges
Health Advisory & Recovery Team | December 24, 2022
The FDA have finally conceded that the mRNA vaccinations increase the risk of pulmonary embolism. The study that led to this conclusion had a very odd methodology. Using this same methodology other risks were dismissed in an unjustified way. There are two FDA studies which use this same methodology. The first reported on people vaccinated aged 12-64 years and the more recent publication was for those aged 65 years and over. The study on the younger population used insurance databases whereas for the older population it was a medicare database.The studies are repeatedly described as being “rapid” and even “near-real time monitoring” even though nearly two years had passed before they were published.
Imagine you want to see if the risk of a certain condition was higher after vaccination was introduced compared to before. Data from voluntary reporting systems can act as an alarm signal but an accurate measure is better derived from comparing how common the condition is to how common it was in the past.
Ideally, the total number of people diagnosed with the condition for a period after vaccination would be compared to a similar period in a previous year. The MHRA set out to carry out such analysis once a week because of the unprecedented nature and size of the rollout. They have not published any findings.
The FDA chose to only look for 28 days for most conditions. The tally was compared to a 28 day period pre-covid. No justification is given for this short window. For comparison, when the Pandemrix vaccine was rushed out for swine flu in 2009, there was an 8 month lag between vaccination and onset of narcolepsy that was so devastating for the young people affected. (Narcolepsy is a condition where there is sudden paralysis or sleep meaning that a normal life is not possible). It is now known that the spike protein circulates for at least 4 months after vaccination and there has been a post mortem study showing characteristic vaccine induced inflammation of the coronary arteries leading to death 4 months after the last dose. The FDA only looked at the risk for the first 28 days.
Let’s say there was a condition where there were more cases in that 28 day period than in a random 28 day period used as a control. This would be very concerning because of a phenomenon called the “healthy vaccinee effect”. People tend to postpone vaccination when they are unwell such that every condition would be expected to be less common in the period immediately after vaccination. To measure the size of this effect it is important to include conditions in the analysis for which there is no expectation of an association as a control. For example, this group included coeliac disease as a control diagnosis. The FDA did not include a control condition.
The FDA methodology did not make a straightforward comparison. Instead they removed all the people who did have the condition but had not got a complete medical record for 365 days prior to the condition being noted. This might have been more justifiable if they had also removed people from the historical data who had not had an intact record for 365 days, but they did not.
They then removed all the people with the condition after the vaccine who had had a diagnosis of that condition in the 365 days prior to vaccination. They did not do the same for the control group.
If vaccination causes an increased risk of myocardial infarction, pulmonary embolism or other clotting problems then it would not be unreasonable to suspect that people who already had a propensity for those conditions would be at highest risk. Not only was their data removed but no separate analysis was reported for this group.
Having removed these people from the vaccination group the scales tipped such that there was only a slight difference between the control group (including people with bad records and previous history) and the post vaccination group (excluding those people). Even then the risk after vaccination was still higher in most of the groups analysed than in the control group.
The FDA then compared the vaccination group with data on people who had had an influenza vaccination. Again, they do not state that they excluded people who had not got an intact medical record or who had had that condition recently. Finally, they managed to tip the scale enough to claim that the vaccines were safe for myocardial infarctions and clotting issues but not pulmonary embolisms in the over 65 year olds. The rate of transverse myelitis was 4 to 7 times higher in those 12-64 year olds given Janssen (an adenovirus vaccine similar to Astrazeneca) but the FDA still concluded they “identified no safety signals.” … Full article
Opposition to Childhood Vaccine Mandates on the Rise, More Parents Say They Want the Right to Choose
Michael Nevradakis, Ph.D. – The Defender – December 16, 2022
A growing number of parents oppose vaccine mandates as a precondition for public school attendance, and interest among adults in receiving COVID-19 booster shots is waning, according to a national poll by the Kaiser Family Foundation (KFF).
The results of the latest KFF COVID-19 Vaccine Monitor survey, released today, show more than one-third (35%) of parents now believe they should be the ones to decide whether their children receive a slate of childhood vaccines.
The poll encompassed a nationally representative sample of 1,259 adults who were interviewed between Nov. 29 and Dec. 8. According to The New York Times, the KFF is a “nonpartisan health care research organization.”
“It’s unfortunate that it took a wave of injuries and deaths from vaccines that never should have been released into the market — much less mandated — to draw long-overdue attention to the issue of vaccine safety,” said Robert F. Kennedy, Jr., chairman and chief litigation counsel for Children’s Health Defense.
Kennedy told The Defender :
“This latest poll is encouraging for those parents, physicians and scientists who for decades have been calling for an investigation into the relentless promotion by FDA, CDC and Big Pharma of inferior medical products without rigorous safety testing.
“As more parents begin to question the forced, routine administration of vaccines on healthy children, perhaps we will move closer to protecting children and holding vaccine makers and government agencies accountable for the harm these products cause.”
26% of parents today: ‘Risks of childhood vaccines for measles, mumps, and rubella outweigh the benefits’
According to the KFF poll, 65% of parents of children under age 18 “think healthy children should be required to be vaccinated to attend public schools.”
This represents an 11% decline from an October 2019 Pew Research Center poll showing 76% of parents supported public school vaccine mandates.
More than one-third of parents surveyed (35%) “now believe parents should be able to decide not to vaccinate their children, up from 23% in 2019.”
The poll also revealed declines in support for specific vaccines. For instance, 71% of respondents said “healthy children should be required to get vaccinated for MMR in order to attend public schools” compared with 82% who supported the MMR vaccine mandate for healthy children in 2019.
Nearly 3 in 10 parents (28%) said parents should be able to choose whether their children receive the MMR vaccine, compared with 16% in the 2019 poll.
A similar percentage (26%) responded that the “risks of childhood vaccines for measles, mumps, and rubella outweigh the benefits.”
A smaller decline was noted in the percentage of adults (85%) who felt the benefits of childhood MMR vaccination outweigh the risk. This represented a three-percentage-point decline from the 2019 Pew Research Center poll (88%).
These declines were driven by increased vaccine “skepticism” and a growing movement toward parental choice, on the part of Republicans and Republican-leaning independents — 44% of whom responded that parents should have a choice about whether or not their children receive the MMR vaccine, up from 20% in 2019.
Only 11% of Democrats provided the same response.
Moreover, only 56% of Republicans and Republican-leaning independents said “healthy children should be required to be vaccinated to attend public schools,” a decline of 23 percentage points compared to 2019.
A similar divide was apparent among respondents in reference to their COVID-19 vaccination status. While 83% of vaccinated respondents said healthy children should be required to be vaccinated in order to attend public schools, 63% of unvaccinated parents said parents should instead decide.
‘Tepid’ interest in COVID ‘boosters’ and flu vaccine
Interest in the updated COVID-19 booster is “tepid,” according to the KFF poll, which showed only 1 in 5 adults (22%) surveyed said they have received the updated bivalent booster and an additional 16% said they plan to receive it “as soon as possible.”
However, 12% of respondents said they would “wait and see” before deciding whether to get the new booster, 13% said they would get it only if required and 9% said they would “definitely not” get it.
An additional 27% were unvaccinated or only “partially” vaccinated, which means they are not eligible to get the booster.
Interest in the bivalent booster was highest among adults 65 and older (39%) and Democrat voters (38%), though both figures fall significantly short of a majority. Conversely, only 12% of Republicans and 11% of young adults under 30 said they had received a dose of the updated booster.
Also, 36% of “fully vaccinated” adults 65 and older said they don’t think they need the updated booster, while a “similar percentage,” according to KFF, said they did not think the benefit of the updated booster was worth it.
Overall, fewer than half of parents of children under 18 said their child has received the updated booster or is likely to do so.
Combined with children who have not been vaccinated and who are therefore ineligible for the booster, 58% of parents of 12- to 17-year-olds and 70% of parents of 5- to 11-year-olds responded in this manner.
Republicans and Republican-leaning independents, even if vaccinated, expressed skepticism toward the updated booster, with 64% stating they do not think they need it, and 61% saying they did not believe the benefit was worth it.
Even among Democrats, a majority (51%) said they were too busy or hadn’t had the time to get the updated booster, indicating it was not a high priority for them.
Even in the face of a so-called “tripledemic” of COVID-19, flu and RSV (respiratory syncytial virus) this fall and winter, and despite the majority of parents saying they are worried their children will get sick from RSV (56%, and 73% of parents of children under the age of 5), only 34% of parents said their child has gotten a flu shot this season.
Parents’ rights movement growing in prominence
According to The Times, “The shift in positions appears to be less about rejecting the shots than a growing endorsement of the so-called parents’ rights movement.”
Dr. Sean O’Leary, chairman of the American Academy of Pediatrics’ Committee on Infectious Diseases told The Times :
“The talking point that has been circulated is the concept of taking away parents’ rights. And when you frame it that simply, it’s very appealing to a certain segment of the population.”
O’Leary said he worried that the parental rights movement might slow down compliance with state-mandated childhood immunization schedules, telling The Times “We do have a global dip in vaccine coverage. So this is not a time to be considering a rollback of these laws.”
Michael Nevradakis, Ph.D., based in Athens, Greece, is a senior reporter for The Defender and part of the rotation of hosts for CHD.TV’s “Good Morning CHD.”
This article was originally published by The Defender — Children’s Health Defense’s News & Views Website under Creative Commons license CC BY-NC-ND 4.0. Please consider subscribing to The Defender or donating to Children’s Health Defense.
Featured Video

Iran War Is Accelerating the End of US Dominance
or go to
Aletho News Archives – Video-Images
From the Archives
Why the UK, the EU and the US Gang-Up on Russia
By James Petras :: 03.20.2018
Introduction: For the greater part of a decade the US, the UK and the EU have been carrying out a campaign to undermine and overthrow the Russia government and in particular to oust President Putin. Fundamental issues are at stake including the real possibility of a nuclear war.
The most recent western propaganda campaign and one of the most virulent is the charge launched by the UK regime of Prime Minister Theresa May. The Brits have claimed that Russian secret agents conspired to poison a former Russian double-agent and his daughter in England, threatening the sovereignty and safety of the British people. No evidence has ever been presented. Instead the UK expelled Russian diplomats and demands harsher sanctions, to increase tensions. The UK and its US and EU patrons are moving toward a break in relations and a military build-up.
A number of fundamental questions arise regarding the origins and growing intensity of this anti-Russian animus.
Why do the Western regimes now feel Russia is a greater threat then in the past? Do they believe Russia is more vulnerable to Western threats or attacks? Why do the Western military leaders seek to undermine Russia’s defenses? Do the US economic elites believe it is possible to provoke an economic crisis and the demise of President Putin’s government? What is the strategic goal of Western policymakers? Why has the UK regime taken the lead in the anti-Russian crusade via the fake toxin accusations at this time?
This paper is directed at providing key elements to address these questions. … continue
Blog Roll
 Aletho News
Aletho News- Pentagon Fast Tracks Iran War Ground Option
- Angela Lipps Spent 108 Days in Jail Because a Facial Recognition Algorithm Was Wrong
- Trump White House plagiarized Iran war manifesto from Israel-aligned think tank
- Western media whitewashes Israel’s attempted murder of British journalist in Lebanon
- Israel-linked arms facility set ablaze in EU
- IRAN: The Three Islands Unmaking American Unipolar Sea Power in the Persian Gulf
- Trump’s dispatch of Marine Expeditionary Unit signals desperation for any symbolic success
- Iran Proposes Creating Middle East Security Structure With All Regional States – President
- Iran’s weapons industry still churning out missiles despite war: IRGC
- Washington approves billions in new arms sales to Gulf states as concerns grow over stocks of air defenses
- If Americans Knew
- Meet the former fashion blogger and shady doctor behind the ‘30,000 dead’ Iran psy-op
- Vatican Secretary of State to Trump, Israel: End the war as soon as possible
- The Majority of Americans Believe War Against Iran Benefits Israel More Than US
- Efforts to shut down pro-Palestinian speech face series of setbacks in court
- What has Israel done to my brother?
- Eid without worship in Al Aqsa – Not a ceasefire Day 161
- Trump: Israel attacked Iranian gas field without US knowledge. No more such attacks!
- America’s friends must help extricate it from an unlawful war
- Democratic Nat’l Committee’s Kneejerk Backing of Israel Is Political and Moral Failure
- Major revelations from former counterterrorism chief Joe Kent’s extensive interview
- No Tricks Zone
- Energy Expert: Germany’s Nuclear Phaseout Was A “500 Billion Euro Mistake”
- New Research: South Australia’s Mid-Holocene Sea Surface Temperatures Were 4°C Warmer Than Today
- Storing Green Energy To Last Germany 10 Days Would Require A 60-Million Tonne Battery
- New Studies: UK Sea Levels Were 4 Meters Higher Than Today During The Mid-Holocene
- Destructive Green New Deal: German Energy And Metal Group Warns Of Drastic Crisis
- New Study Documents A 20-Year Pause In Arctic Sea Ice Decline – Driven By Internal Variability
- Wake-up Call: Survey Shows Majority Of Germans Now Favor Postponing Climate Targets!
- Televised! Leading German Political Candidate Tells Schoolchildren CO2 Makes Sun Hotter!
- New Study: A Century Warming Of 1.1°C Is ‘Commonplace’ And ‘Not Unusual’ During This Interglacial
- New Study: ‘Internal Noise’ And Volcanic Forcing Can Trigger 10-15°C Warming Within Decades

{kind=link}