Critics Slam Fluoride Study by Researchers With Ties to Pro-Fluoride Lobby
By Brenda Baletti, Ph.D. | The Defender | June 2, 2025
Mainstream media outlets are touting a study published May 30 in JAMA Health Forum that predicts ending water fluoridation will worsen children’s oral health and increase national dental healthcare costs.
The study is the most recent attempt by researchers with links to pro-fluoridation lobbying groups like the American Dental Association (ADA) to undermine public confidence in the growing body of scientific evidence that water fluoridation has negative consequences for children’s health.
The study was published by Harvard’s Sung Eun Choi, Ph.D., and Brigham and Women’s Hospital’s Lisa Simon, M.D., D.M.D. Simon receives funding for other research from the ADA, the California Dental Association and other pro-water fluoridation groups, according to the study’s conflict-of-interest disclosures.
Choi and Simon estimated that If everywhere else in the U.S. were to stop fluoridating water, in the next five years, 7.5% more U.S. children ages 0-19 would get cavities — a total of 25.4 million additional teeth would be affected — and it would cost a total of $9.8 billion to treat them.
They also claimed that the number of cavities would more than double in 10 years, to 53.8 million.
The authors argue in the paper and in the press that stopping water fluoridation would disproportionately affect low-income children who are often on Medicaid or without insurance.
Leading fluoride expert Kathy Thiessen, Ph.D., told The Defender there is no good evidence that water fluoridation helps low-income people — it’s just “wishful thinking,” she said, used to justify water fluoridation.
She added:
“Caries development is probably far more related to diet (e.g., sugar) and nutrition (adequate calcium, protein, vitamins) than to fluoride or dental hygiene. That generally translates to higher income, better dental health; lower income, worse dental health.
“The U.S. would be much better off if the money spent on promoting and implementing fluoridation were spent on providing dental care, nutrition, etc., for the lower socioeconomic groups.”
The study authors acknowledged the recent research showing that fluoride exposure has serious negative consequences for children’s neurodevelopment. However, they said that because current federal guidelines haven’t changed to account for such damage, they didn’t consider it in their model.
They didn’t mention that the U.S. Environmental Protection Agency (EPA) is under a court order, which it has yet to appeal, to revise its water fluoridation regulations to account for this risk to children.
Instead, they cited editorials published by members of the ADA and its National Fluoride Advisory Committee, challenging two studies on fluoride’s neurotoxicity to downplay their importance.
Experts on fluoride’s neurotoxic effects who spoke with The Defender were highly critical of the study’s failure to consider fluoride’s neurotoxic effects on children.
Dr. Hardy Limeback, former head of preventive dentistry at the University of Toronto and a fluoride expert said, “Banning fluoridation is a step closer to children’s overall health.”
“Why damage 75 million kids’ brains or the appearance of 9 million kids’ smiles, just to try and save maybe 25 million teeth — if that’s even close to a reliable number — from dental decay?” he asked.
Theissen said the study’s authors don’t include any of the significant costs that result from fluoride’s neurotoxic effects — ranging from immediate healthcare costs, to costs of therapy for disorders such as autism or ADHD, to lifelong earnings reductions associated with lowered IQ.
“A responsible cost-effectiveness analysis really should have included cognitive effects and other adverse effects,” she said.
Fluoride added to drinking water a byproduct of phosphate fertilizer production
Fluoride has been added to community water supplies in the U.S since the 1940s, on the assumption that it would improve children’s dental health.
For decades, scientists and community activists have been raising concerns that fluoride is linked to reduced IQ, behavioral issues, disruption of thyroid functioning and disruption of the gut microbiome.
However, it wasn’t until consumer advocacy groups who sued the EPA in federal court to end water fluoridation won their landmark lawsuit last year that the issue generated national attention.
Soon after Judge Edward Chen ruled that water fluoridation at current U.S. levels poses an “unreasonable risk” of reduced IQ in children and that the EPA must take regulatory action, numerous communities across the country organized campaigns to stop fluoridating their water.
Although most media reports highlight that fluoride is a “naturally occurring mineral,” the fluoride added to water supplies is not.
The fluoride most commonly added to U.S. drinking water supplies is hydrofluorosilicic acid, the byproduct of phosphate fertilizer production, sold off by chemical companies to local water departments across the country.
Overwhelming scientific research shows that fluoride’s benefits to teeth are topical, not the result of ingesting fluoride, and a 2024 Cochrane Review found adding fluoride to drinking water provides very limited dental benefits, especially compared with 50 years ago.
Experts question new study’s model, assumptions and ‘sloppy’ errors
Thiessen called the new JAMA paper “somewhat sloppy,” and cited several outright errors she said reviewers should have caught. She pointed out that the authors confused the roles of different regulatory agencies, provided incorrect citations for some of their model input numbers, and sometimes used outdated cost estimates.
To estimate the effects of ending water fluoridation, the authors created a nationally representative sample using data from 8,484 children, from birth through age 19. The data came from the National Health and Nutrition Examination Survey, which is conducted each year by the CDC and is based on interviews about diet and details from people’s health records.
The study authors used current water fluoridation levels as a proxy for how much fluoride children are exposed to, then predicted the increase in cavities that would occur if that were to stop.
Their model predicted two scenarios: if every public water system fluoridated its water at today’s recommended level of 0.7 milligrams per liter, and if there were a total national ban.
Experts questioned the use of fluoride in water as a proxy for exposure, given that children are exposed to fluoride from many sources other than drinking water, including toothpaste and all processed foods and drinks made with fluoridated water.
They also criticized the “total ban” scenario, in which the researchers estimated that fluoride levels would be reduced to zero in all systems. According to the CDC, almost all water contains some naturally occurring fluoride, so the zero fluoride estimate scenario can’t occur.
It was also “assumed” that children benefit from drinking fluoridated water, but Thiessen said there is no basis for this assumption.
“We badly need some honest and thorough evaluation of whether there is a benefit or not from fluoride or fluoridation,” she said. “If there is no real benefit, then obviously any risk of adverse health effects is not justified.”
The only negative health effect of water fluoridation the researchers considered was dental fluorosis — a discoloration of the teeth that occurs when a child is overexposed to fluoride.
Even their estimate of how many children would have “objectionable” dental fluorosis “completely missed the mark,” Limeback said. According to the Cochrane Review cited by the researchers, every eighth child in fluoridated areas has dental fluorosis that needs repair, Limeback said. Ending fluoridation would result in 9,375,000 (not 200,000 as they reported) fewer cases of dental fluorosis.
Each case of serious fluorosis costs between $2,000 and $20,000 to repair, he said, meaning that ending fluoridation offers potential savings of $18.75 to $187.5 billion dollars.
“America would drastically reduce the dental fluorosis epidemic in the U.S. if all the states banned water fluoridation.”
Thiessen also noted that the authors disregarded other costs borne by the American public associated with water fluoridation, including the costs of fluoridating, and the costs of cleaning up fluoridation overfeeds and spills, which are common, and addressing the health issues they cause.
“I also expect that other health issues will decrease substantially, more than making up for any increase in dental costs,” Thiessen added.
This article was originally published by The Defender — Children’s Health Defense’s News & Views Website under Creative Commons license CC BY-NC-ND 4.0. Please consider subscribing to The Defender or donating to Children’s Health Defense.
FDA exposed – hundreds of drugs approved with no proof they work
Two-year investigation reveals a broken approval system, ineffective—and sometimes deadly—drugs fast-tracked to market without evidence
By Maryanne Demasi, PhD | June 5, 2025
The US Food and Drug Administration (FDA) has approved hundreds of drugs without proof they work—and in some cases, despite evidence they cause harm.
That’s the finding of a blistering two-year investigation by medical journalists Jeanne Lenzer and Shannon Brownlee, published by The Lever.
Reviewing more than 400 drug approvals between 2013 and 2022, the authors found the agency repeatedly ignored its own scientific standards.
One expert put it bluntly—the FDA’s threshold for evidence “can’t go any lower because it’s already in the dirt.”
A system built on weak evidence
The findings were damning—73% of drugs approved by the FDA during the study period failed to meet all four basic criteria for demonstrating “substantial evidence” of effectiveness.
Those four criteria—presence of a control group, replication in two well-conducted trials, blinding of participants and investigators, and the use of clinical endpoints like symptom relief or extended survival—are supposed to be the bedrock of drug evaluation.
Yet only 28% of drugs met all four criteria—40 drugs met none.
These aren’t obscure technicalities—they are the most basic safeguards to protect patients from ineffective or dangerous treatments.
But under political and industry pressure, the FDA has increasingly abandoned them in favour of speed and so-called “regulatory flexibility”.
Since the early 1990s, the agency has relied heavily on expedited pathways that fast-track drugs to market.
In theory, this balances urgency with scientific rigour. In practice, it has flipped the process. Companies can now get drugs approved before proving they work, with the promise of follow-up trials later.
But, as Lenzer and Brownlee revealed, “Nearly half of the required follow-up studies are never completed—and those that are often fail to show the drugs work, even while they remain on the market.”
“This represents a seismic shift in FDA regulation that has been quietly accomplished with virtually no awareness by doctors or the public,” they added.
More than half the approvals examined relied on preliminary data—not solid evidence that patients lived longer, felt better, or functioned more effectively.
And even when follow-up studies are conducted, many rely on the same flawed surrogate measures rather than hard clinical outcomes.
The result: a regulatory system where the FDA no longer acts as a gatekeeper—but as a passive observer.
Cancer drugs: high stakes, low standards
Nowhere is this failure more visible than in oncology.
Only 3 out of 123 cancer drugs approved between 2013 and 2022, met all four of the FDA’s basic scientific standards.
Most—81%—were approved based on surrogate endpoints like tumour shrinkage, without any evidence they improved survival or quality of life.
Take Copiktra, for example—a drug approved in 2018 for blood cancers. The FDA gave it the green light based on improved “progression-free survival,” a measure of how long a tumour stays stable.
But a review of post-marketing data showed that patients taking Copiktra died 11 months earlier than those on a comparator drug.
It took six years after those studies showed the drug reduced patients’ survival for the FDA to warn the public that Copiktra should not be used as a first- or second-line treatment for certain types of leukaemia and lymphoma, citing “an increased risk of treatment-related mortality.”
Elmiron: ineffective, dangerous—and still on the market
Another striking case is Elmiron, approved in 1996 for interstitial cystitis—a painful bladder condition.
The FDA authorised it based on “close to zero data,” on the condition that the company conduct a follow-up study to determine whether it actually worked.
That study wasn’t completed for 18 years—and when it was, it showed Elmiron was no better than placebo.
In the meantime, hundreds of patients suffered vision loss or blindness. Others were hospitalised with colitis. Some died.
Yet Elmiron is still on the market today. Doctors continue to prescribe it.
“Hundreds of thousands of patients have been exposed to the drug, and the American Urological Association lists it as the only FDA-approved medication for interstitial cystitis,” Lenzer and Brownlee reported.
“Dangling approvals” and regulatory paralysis
The FDA even has a term—”dangling approvals”—for drugs that remain on the market despite failed or missing follow-up trials.
One notorious case is Avastin, approved in 2008 for metastatic breast cancer.
It was fast-tracked, again, based on ‘progression-free survival.’ But after five clinical trials showed no improvement in overall survival—and raised serious safety concerns—the FDA moved to revoke its approval for metastatic breast cancer.
The backlash was intense.
Drug companies and patient advocacy groups launched a campaign to keep Avastin on the market. FDA staff received violent threats. Police were posted outside the agency’s building.
The fallout was so severe that for more than two decades afterwards, the FDA did not initiate another involuntary drug withdrawal in the face of industry opposition.
Billions wasted, thousands harmed
Between 2018 and 2021, US taxpayers—through Medicare and Medicaid—paid US$18 billion for drugs approved under the condition that follow-up studies would be conducted. Many never were.
The cost in lives is even higher.
A 2015 study found that 86% of cancer drugs approved between 2008 and 2012 based on surrogate outcomes showed no evidence they helped patients live longer.
An estimated 128,000 Americans die each year from the effects of properly prescribed medications—excluding opioid overdoses. That’s more than all deaths from illegal drugs combined.
A 2024 analysis by Danish physician Peter Gøtzsche found that adverse effects from prescription medicines now rank among the top three causes of death globally.
Doctors misled by the drug labels
Despite the scale of the problem, most patients—and most doctors—have no idea.
A 2016 survey published in JAMA asked practising physicians a simple question —what does FDA approval actually mean?
Only 6% got it right.
The rest assumed it meant the drug had shown clear, clinically meaningful benefits—such as helping patients live longer or feel better—and that the data was statistically sound.
But the FDA requires none of that.
Drugs can be approved based on a single small study, a surrogate endpoint, or marginal statistical findings. Labels are often based on limited data, yet many doctors take them at face value.
Harvard researcher Aaron Kesselheim, who led the survey, said the results were “disappointing, but not entirely surprising,” noting that few doctors are taught about how the FDA’s regulatory process actually works.
Instead, physicians often rely on labels, marketing, or assumptions—believing that if the FDA has authorised a drug, it must be both safe and effective.
But as The Lever investigation shows, that is not a safe assumption.
And without that knowledge, even well-meaning physicians may prescribe drugs that do little good—and cause real harm.
Who is the FDA working for?
In interviews with more than 100 experts, patients, and former regulators, Lenzer and Brownlee found widespread concern that the FDA has lost its way.
Many pointed to the agency’s dependence on industry money. A BMJ investigation in 2022 found that user fees now fund two-thirds of the FDA’s drug review budget—raising serious questions about independence.
Yale physician and regulatory expert Reshma Ramachandran said the system is in urgent need of reform.
“We need an agency that’s independent from the industry it regulates and that uses high quality science to assess the safety and efficacy of new drugs,” she told The Lever. “Without that, we might as well go back to the days of snake oil and patent medicines.”
For now, patients remain unwitting participants in a vast, unspoken experiment—taking drugs that may never have been properly tested, trusting a regulator that too often fails to protect them.
And as Lenzer and Brownlee conclude, that trust is increasingly misplaced.
- Investigative report by Jeanne Lenzer and Shannon Brownlee at The Lever [link]
- Searchable public drug approval database [link]
- See my talk: Failure of Drug Regulation: Declining standards and institutional corruption
Australia quietly pivots on Covid-19 vaccine policy
By Maryanne Demasi, PhD | June 3, 2025
It didn’t come with a press conference or a media blitz. In fact, there was no announcement at all.
But sometime around 2 May 2025, the Australian Department of Health quietly removed its recommendation for Covid-19 vaccination in healthy children and adolescents under 18.

The change was tucked into an online update to the Australian Immunisation Handbook—no headline, no ministerial statement, no media campaign to inform the public.
For the first time since the rollout began, Australian health authorities now say that unless a child has underlying medical conditions, they do not need the vaccine.
Australia now joins a growing list of countries backing away from the blanket approach to vaccinating low-risk populations.
In the US, health officials under HHS Secretary Robert F. Kennedy Jr. recently removed routine recommendations for Covid-19 vaccination in healthy children and pregnant women.
The CDC now leaves it up to “shared decision-making”—a tacit acknowledgment that the previous universal approach may have overreached.
Denmark, meanwhile, was ahead of the curve.
It stopped recommending the vaccine for healthy children back in 2022, citing data showing that severe Covid in children was exceedingly rare and that the benefits of mass vaccination did not outweigh the harms.
Australia’s policy reversal might be late, but what makes it striking is how quietly it was done—and how much it implicitly concedes.
For years, anyone who questioned the need to vaccinate healthy children was dismissed as anti-science or dangerous. Now, the same authorities who widely promoted the shots are quietly walking it back.
And the adverse events that critics raised early on—myocarditis, pericarditis, and other post-vaccine complications—are no longer fringe concerns. They’re acknowledged in official risk assessments.
The shift also comes at a time when the legal and regulatory framework that enabled the rapid approval of mRNA vaccines is under growing scrutiny.
In Australia, a case brought by Dr Julian Fidge, a general practitioner and former pharmacist, challenged the legality of the vaccine approvals.
He argued that Pfizer and Moderna’s mRNA vaccines should have been classified as “genetically modified organisms” under the Gene Technology Act 2000, and therefore required a licence from the Office of the Gene Technology Regulator (OGTR) before being rolled out.
But the court dismissed the case on procedural grounds, ruling that Dr Fidge lacked standing to pursue it.
Still, the case drew attention to whether these products were channelled through the wrong regulatory pathway.
That question is now at the centre of a citizen petition in the US, filed with the FDA in January 2025, claiming the agency “wrongfully and illegally” approved the mRNA Covid-19 vaccines by treating them as conventional biologics, not gene therapies.
According to the FDA’s own definition, gene therapy products are those that use genetic material to alter cellular function for therapeutic use.
By that logic, mRNA vaccines clearly qualify – and should have faced far more rigorous safety testing, including environmental risk assessments and long-term follow-up studies.
As of June, the FDA has not responded to the petition—but the implications are enormous.
If regulators in Australia or the US misclassified these products during the emergency rush, it would expose a systemic failure to apply the appropriate safeguards to an entirely new class of biotechnology.
And it’s not just about legal definitions. The public mood is shifting.
The notion that healthy children and adolescents should have been part of a sweeping global experiment with novel gene-based technologies now looks reckless in hindsight. For the public, trust has been damaged—perhaps irreparably.
That shift in perception has consequences far beyond Covid.
Billions of dollars have been invested in mRNA platforms for other diseases—flu, RSV, and cancer. So what happens if confidence in the technology craters?
Already, the US FDA has announced it will require new randomised clinical trials for annual Covid-19 boosters in “healthy” people under 65—setting a higher threshold for evidence (than immunobridging data) that may make future approvals more challenging.
The industry might dismiss this as just a hiccup—but the truth is, mRNA vaccines were never subjected to the kind of long-term scrutiny typically required of products given to healthy people, especially children.
The argument that urgency justified shortcuts has worn thin.
The real emergency now is institutional—one of captured regulators, collapsing public trust, and a health system so entangled with the pharmaceutical industry it can no longer tell the difference between evidence and marketing.
Is the FDA a mutinous ship?
By Dr Clare Craig | Health Advisory & Recovery Team | June 3, 2025
They promised change. They promised transparency. They promised reform. But with the FDA’s latest approval of Moderna’s mNEXSPIKE® covid jab, it is clear the only thing that has changed is the branding.
This so-called “reformed” FDA has just authorised a new mRNA vaccine — without testing it against a placebo. Instead, the comparator was Moderna’s previous product, already associated with a range of known adverse effects.
And yet, somehow, we’re meant to believe this is progress.
Meanwhile, the leaders heading up MAHA, elected on a promise to end the regulatory theatre and restore public trust, appear to be captaining a vessel still drifting in the same dangerous waters. They are on a mutinous ship and are wrestling for the wheel.
While the FDA continues to greenlight successive iterations of mRNA vaccines, other major Western nations long since reduced their ambitions:
- In the United Kingdom, the Joint Committee on Vaccination and Immunisation (JCVI) now recommends boosters only for individuals aged 75 and over, residents of care homes, and those with specific clinical vulnerabilities.
- France and Germany have similarly curtailed their booster programmes, focusing on high-risk populations and refraining from broad recommendations for younger, healthier demographics.
This more cautious approach is not justifiable either given the failure of these product and their safety profile. The “benefit” only ever was – and continues to be – a statistical illusion. No one should be being exposed to this unnecessary risk.
This divergence raises a critical question: How can the same body of evidence lead to such different public health policies?
Latest Inadequate Trials
The Phase 3 trial underpinning mNEXSPIKE’s approval enrolled approximately 11,400 participants aged 12 and older. While this number might seem substantial, it’s important to note that only about half received the mNEXSPIKE vaccine, while the remainder received an earlier Moderna product as a comparator. There was no placebo comparison.
This sample size is insufficient to detect rare but serious adverse events – even with a placebo. Without one you can only see differences between two sets of harm.
For context, previous studies have indicated that mRNA vaccines may be associated with an excess risk of serious adverse events of special interest, estimated at approximately 15.1 per 10,000 vaccinated individuals. This is totally unacceptable for general use even in a product that has significant benefits.
Despite approval, critical safety studies are still pending. One study, assessing safety in pregnant women, is not due until 2032. Another, evaluating vaccine effectiveness for adults aged 50–64, is still in the planning stages.
Are the FDA still pretending there is an emergency to justify these rushed decisions?
A Hollow Reformation
The USA public deserves better from their officials. They are paying for this reckless approach. The question is, can those with a more precautionary, less ideological approach, wrestle the wheel of the ship and steer her to safety?
Covid-19 vaccine reform is moving slower than many had hoped
Moderna’s latest mRNA vaccine approval stuns reform advocates—but real change demands persistence when science runs up against powerful interests
By Maryanne Demasi, PhD | June 1, 2025
Just three weeks after Dr Vinay Prasad assumed oversight of vaccines at the FDA, Moderna’s latest Covid-19 vaccine, mNEXSPIKE®, received full approval.
For those who had hoped the mRNA platform would be shelved, the decision landed like a gut punch.
Approved on 31 May 2025, the next-generation shot is intended for adults over 65, as well as individuals aged 12 to 64 with at least one risk factor for severe illness.
And it came under the watch of a man who had spent years demanding greater scientific rigour from the agency.
Prasad had been among the FDA’s most outspoken critics during the pandemic, repeatedly condemning its reliance on surrogate endpoints—such as antibody levels—rather than hard clinical outcomes like reduced hospitalisation or death.
And he didn’t just say it once. He drove the point home, over and over.
“Showing boosters improve neutralizing antibodies or other laboratory measures is not what we need,” he posted on X in July 2022. “We need randomized control trials powered for clinical endpoints showing boosters improve outcomes that people care about.”
In January 2023, he co-signed a formal Citizen Petition to the FDA stating, “This immunobridging surrogate endpoint has not been validated to predict clinical efficacy.”
Then in March 2023, he made his position even clearer on Substack. “I don’t care about transient antibody titer levels,” he wrote.
But mNEXSPIKE® appears to have been approved primarily using exactly those kinds of data—measures of immune response, not measures of meaningful outcomes.
So how do we square that?
Technically, the approval aligns with the policy Prasad outlined in a recent New England Journal of Medicine article.
There, he proposed a two-track system — no further vaccine approvals for healthy adults without RCTs showing clinical benefit—but for older adults and at-risk individuals, immunobridging data could still be acceptable.
So yes, by that standard, mNEXSPIKE® fits the rules.
But it doesn’t erase the discomfort. Because for years, Prasad insisted those very shortcuts—approving Covid vaccines based on antibody levels instead of clinical outcomes—were scientifically flimsy.
Now, under his watch, those same shortcuts are back in play.
When Robert F. Kennedy Jr. was appointed HHS Secretary, reform didn’t just seem likely—it felt imminent.
Many expected the mRNA shots would be pulled from the market, or at the very least, that new approvals would be frozen until stronger evidence emerged.
Instead, we’ve seen a flood of high-production videos and polished slogans about “restoring public trust.”
To many observers, it looks like transparency on the surface—but business as usual underneath.
Of course, no one said this would be easy.
Having worked in government as a political adviser, I know how hard it is to shift systems that are not only slow and bureaucratic, but deeply enmeshed with commercial interests. And no sector is more heavily invested in mRNA than biotech.
This isn’t just about Covid anymore. The pharmaceutical industry has poured billions into mRNA vaccines for RSV, flu, HIV, cancer, and more. Entire product pipelines are now staked on the assumption that the technology is here to stay.
Pulling the plug wouldn’t just alter public health policy—it would tank portfolios, gut R&D budgets, and unleash a political and financial firestorm from some of the most powerful corporate interests on earth.
That’s the kind of pressure Prasad is under now. That’s the reality Kennedy’s team has stepped into.
This is no longer science versus ideology. It’s science versus entrenched industry power.
And many are beginning to worry we’re watching the same playbook unfold—just with better branding.
That’s not what MAHA supporters or vaccine-injured families were hoping for. They’re not asking for tweaks. They want the shots gone. Not revised. Not updated—just gone.
But political reality rarely keeps pace with public demand. Even the most determined reformers can’t move faster than the machinery they’re trying to dismantle.
So where does that leave us?
Facing the hardest task of all—staying in the fight.
Progress may feel glacial, but it is underway.
The CDC has removed routine Covid-19 vaccine recommendations for healthy children and pregnant women. Prasad’s new framework has halted low-risk approvals unless backed by RCTs.
Yes, the mRNA platform is still alive—and still fiercely protected—but reform was never going to be easy. And it was never going to come all at once.
COVID Doubts Made You a ‘Violent Extremist’
By Jim Bovard | The Libertarian Institute | June 2, 2025
Biden administration policymakers hated you more than you knew.
Four years ago, I warned at the Libertarian Institute:
“Libertarians are in the federal crosshairs… Many libertarians assume they have nothing to fear because they are not engaged in seeking to violently overthrow the government. But the feds will be able to find many other pretexts to target peaceful citizens with supposedly subversive ideas.”
Three years ago, I warned at the Institute that White House Press Secretary Karine Jean-Pierre was damning anyone who did not kowtow to the regime:
“’When you are not with what majority of Americans are, then you know, that is extreme. That is an extreme way of thinking.’ That wacko definition of extremism designed to vilify anyone who doubts Biden will save America’s soul.”
In October 2023, I warned at the Institute:
“Federal bureaucrats heaved together a bunch of letters to contrive an ominous new acronym for the latest peril to domestic tranquility. The result: AGAAVE—’anti-government, anti-authority violent extremism’—which looks like a typo for a sugar substitute. The FBI vastly expanded the supposed AGAAVE peril by broadening suspicion from ‘furtherance of ideological agendas’ to ‘furtherance of political and/or social agendas.’ Anyone who has an agenda different from Team Biden’s could be AGAAVE’d for his own good.”
Director of National Intelligence Tulsi Gabbard recently declassified a December 13, 2021 report by the National Counterterrorism Center. Gabbard’s version had a more honest title than the original version: “Declassified Biden Administration Documents Labeling COVID Dissenters, Others as ‘Domestic Violent Extremists.’”
President Joe Biden’s Brain Trust sounded the alarm on criticisms such as “COVID-19 vaccines are unsafe, especially for children, are part of a government or global conspiracy to deprive individuals of their civil liberties and livelihoods, or are designed to start a new social or political order.” After government lockdowns had destroyed millions of jobs, only the paranoid would fear the government would ever violate their liberties or subvert their livelihoods.
Biden policymakers pretended that the surge in criticism of COVID policies was proof of the psychopathology of Biden’s opponents. But in September 2021, Biden dictated that one-hundred million Americans working for private companies must get the COVID vaccine. The official counterterrorism report stated that it anticipated that “the threat will continue at least into the winter, as many of the new COVID-19 mandates in the U.S….are implemented, including U.S. workplace vaccination policies that carry disciplinary or termination penalties.” The Supreme Court struck down most of that vaccine mandate as illegal in January 2022 but not before it had profoundly disrupted legions of lives and businesses—as well as American health care.
The other factor spurring the surge in COVID criticism was the failure of the COVID vaccines. In early 2022, the effectiveness of the COVID booster shot had fallen to 31%—too low to have been approved by the Food and Drug Administration. Though most American adults had gotten COVID vaccines, there were more than a million new COVID cases a day in January 2022. Most COVID fatalities were occurring among the fully vaxxed. Studies showed that people who received multiple boosters were actually more likely to be hit by COVID infections.
So obviously, the Biden administration had no choice but to demonize any and all COVID critics. A confidential 2022 Department of Homeland Security report detailed pending crackdowns on “inaccurate” information on “the efficacy of COVID-19 vaccines,” among other targets. A few months earlier, Jen Easterly, the chief of the Cybersecurity and Infrastructure Security Agency, declared, “We live in a world where people talk about alternative facts, post-truth, which I think is really, really dangerous if people get to pick their own facts.” Plenty of Biden administration officials considered it “really dangerous” to permit people to assert that COVID vaccines were failing.
The National Counterterrorism Center report noted, “The availability of a vaccine for all school-age children might spur conspiracy theories and perceptions that schools will vaccinate children against parents’ will.” Like the same way that some states and many school systems have sought to enable children to change their gender without their parents’ knowledge or consent?
The report also warned that “new COVID-19 mitigation measures—particularly mandates or endorsements of vaccines for children—will probably spur plotting against the government.” The FDA knew that COVID vaccines sharply increased the risk of myocarditis—an inflamed heart—in young males but the Biden White House browbeat the agency into fully approving the COVID vaccine anyhow. New York Governor Kathy Hochul sought unsuccessfully to mandate vaccines for all schoolkids in the Empire State even though her State Department of Health reported in May 2022 that the Pfizer vaccine was only 12% effective for children during the Omicron surge. The Biden administration included COVID vaccines in the semi-mandatory regimen for young children despite the vaccine’s failure and perils.
The vilification of COVID doubts propelled the Biden crackdown on uppity parents. As governments shut down schools and issued mask mandates in failed responses to COVID, parents raised hell at school board meetings. The National School Board Association denounced such criticism as “a form of domestic terrorism” and urged Team Biden to deploy the FBI and the Patriot Act against protesting parents (an initial draft of the letter called for sending in the National Guard to protect school boards).
On October 4, 2021, Attorney General Merrick Garland announced that the FBI would speedily “convene meetings” in every state aimed at “addressing threats against school administrators, board members, teachers, and staff.” The Justice Department announced that its National Security Division would help determine “how federal enforcement tools can be used” to prosecute angry parents. The Biden administration effectively announced plans to drop legal nuclear bombs on school board critics. An FBI whistleblower revealed that FBI counterterrorism tools were being used to target angry parents. FBI agents across the nation began interrogating parents whose names were reported on a “tip line” set up for people to phone in accusations against anyone who complained about school closures, mask mandates, or other issues.
Portraying doubts on COVID policy as a warning sign of domestic violent extremism unleashed the FBI to target anybody who howled against mandatory injections or the near-total destruction of their freedom of movement. That December 13, 2021 National Counterterrorism Center report may be only the tip of the iceberg of federal mischief. We may soon learn of far more direct machinations to vilify, undercut, or other stifle COVID critics.
The BBC’s climate science problem
By Andrew Montford | Net Zero Watch | May 20, 2025
You could be forgiven for thinking the BBC is out to get Richard Tice. Their chosen battleground is the Reform man’s position on climate and Net Zero, but it’s fair to say the campaign is, thus far, not going too well.
Question Time last week was a car crash for the corporation, with chairman Fiona Bruce interrupting Tice to contradict his contention that only 4% of carbon dioxide emissions are manmade. Thirty percent was the correct figure, she boldly asserted. Unfortunately, Tice was right, and she was wrong, so the Corporation’s gophers got to work and quietly edited the recording to remove her gaffe. Unfortunately someone noticed, and sceptics had a field day.
Undeterred, the Corporation returned to the fray a few days later, when Nick Robinson had Tice on his Political Thinking podcast. They decided, somewhat surprisingly, to take up cudgels on exactly the same subject, namely the human influence on climate.
Once again, Tice expained that human emissions were dwarfed by natural ones, and there was no attempt to probe this argument more deeply. The conversation meandered off elsewhere.
However, shortly aferwards Robinson decided to stick in a metaphorical boot, tweeting a clip from the interview with the comment:
He’s denying the scientific consensus that climate change is partly man made & can be slowed or halted.”
This is a very strong take given that Robinson had not attempted to pin down Tice on precisely what he meant. But at face value it’s a misrepresentation.
Tice’s words could only reasonably be interpreted as implying that the human contribution is nugatory compared to the natural one. To get to Robinson’s take – that Tice believed that there was no human influence – would mean considering his words as meaning natural CO2 emissions affected the climate but human ones didn’t. This would be ludicrous.
Tice’s words clearly implied that he thought mankind affected the climate, but only marginally so. In other words, far from “denying… that climate change is partly man made”, this was his starting point!
As to the rest of Robinson’s claim – that Tice was denying that climate change “can be slowed or reversed”, we need to note what appears to be a fatal contradiction in Robinson’s position. If climate change is “partly” manmade, then it is also partly natural. How, we wonder, does Robinson think we can halt the natural element?
It is undoubtedly substantial. We are sure that the climate changes on all timescales, from the decadal and centennial to the millennial and beyond. We know this from, for example, long-term temperature records, such as the Central England Temperature Series, the 800-year record of the waters of the Nile, and proxy climate records covering even longer periods. And the natural changes that are seen in history can be dramatic. One notable example was the sudden temperature rise at the end of the period, over 10,000 years ago, known as the Younger Dryas. Temperatures around the world are thought to have increased by 3–10 degrees in just a few decades.
How does Nick Robinson think we are going to stop that kind of climate change?
Charitably, Robinson – who is a generalist – simply hasn’t thought through what he means by “climate change”. He has no robust understanding of climate history and climate science, and is therefore unable to probe the position of people like Tice, who have given the issues some thought.
That being the case, he needs to think before he speaks, and perhaps to be a little more cautious about dishing out accusations of denial.
How Lies and Hubris Caused an Awakening
By Pat Fidopiastis | Brownstone Institute | May 26, 2025
In March 2020, the phrase “Fifteen days to slow the spread” was transmitting faster than SARS-CoV-2. At the time, it seemed reasonable to want to buy our health care workers a few weeks to prepare. Contemporaneously, Dr. Anthony Fauci reasonably summarized decades of research in his 60 Minutes interview by saying that masks are not an effective way to block respiratory viruses.
In a Snapchat interview, Dr. Fauci reasonably interpreted timely data on Covid-19 outcomes to conclude that young people could decide for themselves if they wanted to meet strangers on a dating app during the pandemic. As Dr. Fauci put it: “Because that’s what’s called relative risk.”
Even the authors of the “proximal origin” opinion piece in Nature Medicine made reasonable points in support of a natural origin of SARS-CoV-2 (despite revealing their cards by calling “lab leak” implausible): “… it is likely that SARS-CoV-2-like viruses with partial or full polybasic cleavage sites will be discovered in other species” and “More scientific data could swing the balance of evidence to favor one hypothesis over another.”
Five years later, thousands of animals have been sampled, millions of genomic sequences have been analyzed, and still there is nothing remotely close to a non-human adapted, animal version of SARS-CoV-2; back in 2003, using “stone tools” compared to today’s technology, they found the animal version of that SARS virus in a few months.
Unfortunately, the honeymoon of reason was brief. Overwhelming evidence that SARS-CoV-2 was not natural became a “destructive conspiracy,” and if you spoke about it, you were somehow racist.
Surgeon General Jerome Adams instructed us on how to make a life-saving mask from an old t-shirt. Dr. Fauci used the bizarre excuse that he lied in his 60 Minutes interview to explain why he abruptly reversed himself and began promoting the epidemiological theater of wearing several masks at once.
Not to be outdone, Dr. Deborah Birx summed up the futility of her leadership with this pearl: “We know that there are ways that you can even play tennis with marked balls so you’re not touching each other’s balls.” This sounded more like a punchline than worthwhile public health advice. Perhaps most egregious of all, we learned that “Two weeks to slow the spread” was not meant to be taken literally.
For me, a professor of microbiology for nearly 25 years, the moment of reason ended when I stepped into an elevator on my campus and saw a floor sticker telling me where to stand (Fig. 1). I simply could not keep quiet and pretend that this was sound public health advice.

Fig. 1
Before long, businesses were inundated with pandemic rules. I was hired by one of the lucky ones deemed “essential,” and therefore allowed to open, to assist with “safe” operation plans.
When I arrived to conduct my inspection, the business looked more like an Ebola field hospital than a furniture store (Fig. 2). Masked customers were herded in the parking lot by ropes and signs. One by one, they were greeted by an attendant, grateful to still have a job, standing behind Plexiglas, wearing a mask and face shield.
The friendly attendant was instructed to ask uncomfortable questions about symptoms like diarrhea. If a customer responded “yes” to any of the symptoms or refused to answer, they could not shop for furniture. If “no,” then their temperature was measured.
It was nearly 100 degrees that day so almost everyone had to be scanned multiple times. Inside the store was a maze of one-way arrows, warning signs, Plexiglas, hand sanitizer stations, and boxes of masks and disposable couch covers. They even had a video monitor reporting the number of customers per 400 square feet of store. Sadly, the epidemiological version of “over-medicating the patient” did not stop with onerous business rules.

Fig. 2
Drunk with power, public health officials in California felt ordained to protect the unwashed masses from Thanksgiving dinner. Unsurprisingly, these farcical dining rules did not apply to everyone.
Who actually believed “singing, chanting, shouting, and physical exertion” at a family dinner was too risky? Who decided that we needed to bulldoze a skate park to prevent kids from congregating? Why was it necessary to arrest a lone paddleboarder in Santa Monica Bay for “flouting coronavirus closures?”
In the LA Times article on the paddler’s arrest, a professor from the prestigious Scripps Institute of Oceanography opined, “SARS-CoV-2, the virus that causes COVID-19, could enter coastal waters and transfer back into the air along the coast. I wouldn’t go in the water if you paid me $1 million right now.”
I tried laughing off the ridiculous, unenforceable Thanksgiving rules, those stickers in the elevators, and other nonsense that at the time was happening somewhere else. But I could not get past the frightening reality that so many of my highly educated peers actually believed nonsense like SARS-CoV-2 was leaping out of the ocean.
Anyone paying attention could compile government data on Covid-19 outcomes and assess risk for themselves (Table 1). The message was always the same – the vast majority of deaths attributed to Covid-19 were people over 65 years old with severe comorbidities, especially obesity.

Table 1
By signing the Great Barrington Declaration and discussing its premise of “focused protection” in my advanced microbiology courses, I received an avalanche of vitriol.
Among the most shocking responses were accusations of “ageism” and “fat-shaming” for discussing hard facts about the pandemic.
Just like that, the “Science doesn’t care about your feelings” crowd started prioritizing their feelings. The university newspaper asked for an interview. I was warned not to accept, but I wanted to start a bigger conversation. I regret my decision because the article they wrote did not represent the views I articulated.
Instead, I was accused of promoting a “power imbalance” by supposedly forcing my “junk science” views on students. I used to think the cries of “fake news” were just a lazy argument by people that could not support their position, until I read that article about me.
Ironically, these same people who attacked me had completely accepted the made-up “six-feet rule,” which was the root of so much collateral damage. Heavily biased news sources like NPR defended this unscientific rule by stating, “distance still protects you.” However, if the cure is not even remotely feasible, despite the best efforts of authoritarians, then it’s not really a cure.
Apparently I crossed the line when I discussed in class how politicized the pandemic had become. How is it that President Trump’s rallies were spreading “coronavirus and death” but BLM protests had no effect on coronavirus cases? The sampling bias was baked in, given that contact tracers were being told not to ask people if they had been to a protest.
Why was it acceptable for CNN to use phrases such as “Wuhan virus” and “Chinese coronavirus,” but when President Trump did it, he was called “racist?” Was it actually “racist” to discuss the obvious signs of genetic manipulation in the SARS-CoV-2 genome with my students in an Emerging Infectious Diseases class?
My campus newspaper and many of my colleagues thought so, as did an Asian American and Pacific Islander group calling for my resignation. When the admonitions about masks became aggressive (Fig. 3), and draconian, unscientific outdoor mask fines were being implemented, I analyzed some data and conducted a few experiments to find out for myself if masks were worth all the anger.

Fig. 3
I looked at “cases” in places like New York City and pointed out when the mask mandate and fines were applied (Fig. 4). Notably, the NYC mandate was instituted after cases had already begun to fall, and coercive fines did not prevent the second wave, which was longer and reached a higher peak than the first wave.
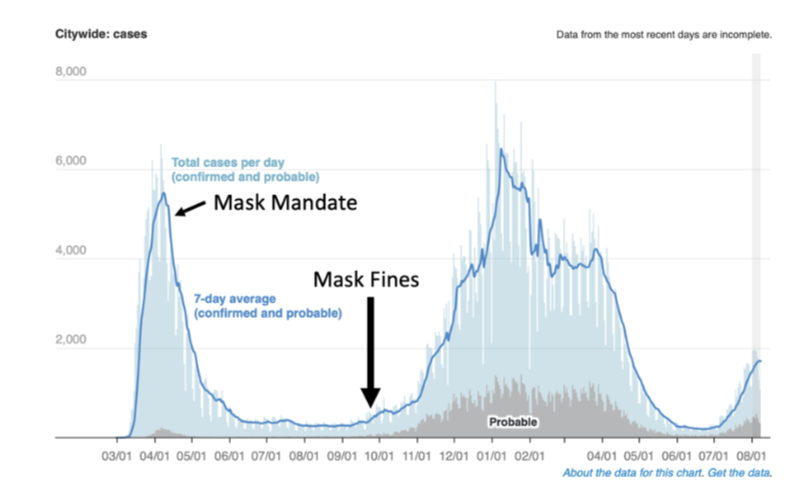
Fig. 4
I had my allergy-prone daughter sneeze onto petri-plates with and without the CDC-approved masks we wore to enter locations that enforced the mask mandate (Fig. 5). The saliva spray patterns, illustrated by microbial growth on the plates, were virtually indistinguishable.
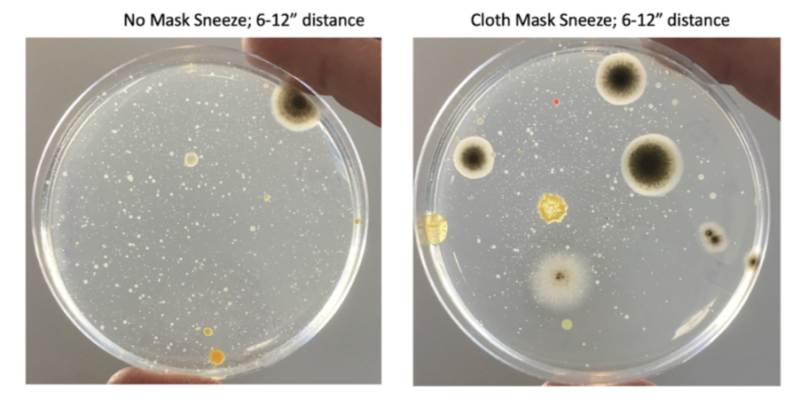
Fig. 5
In the 60 Minutes interview, Dr. Fauci stated that “… often there are unintended consequences…people keep fiddling with the mask and touching their face…” implying that germs collect on masks, making them a source of contagion rather than a barrier.
Indeed, after the sneeze experiment, I stamped the outside of my daughter’s mask onto a petri-plate. The resulting dense microbial growth supported Dr. Fauci’s argument against mask wearing – “fiddling with the mask” probably does spread microbes (Fig. 6).
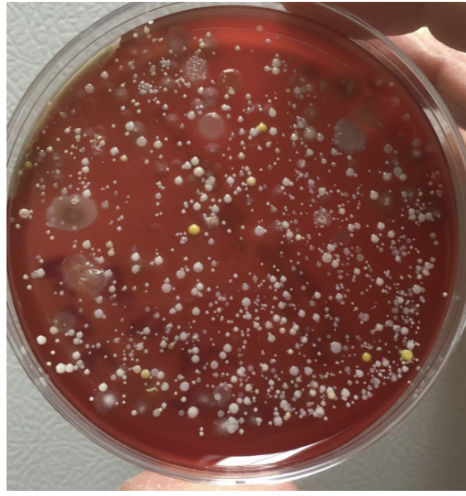
Fig. 6
At the time, I stated in the campus newspaper that “the science on masks was mixed at best.” However, the third-year journalism student apparently knew better and decided I was pushing “junk science.” Was I naïve to expect an apology after “the science” started catching up to what I was saying?
During the pandemic, my lab was responsible for measuring SARS-CoV-2 levels in wastewater (Fig. 7) to use this information as a means of tracking community transmission. We learned two important lessons from this approach.
First, peak levels of SARS-CoV-2 in wastewater (orange line) provided a few weeks’ lead to when we could expect to see peak levels of people testing positive for the virus (i.e., “cases;” blue line). Second, we learned that the mask mandate (red line) did not stop the virus from doing what it wanted. Despite the mask mandate, transmission of SARS-CoV-2 reached unprecedented highs.
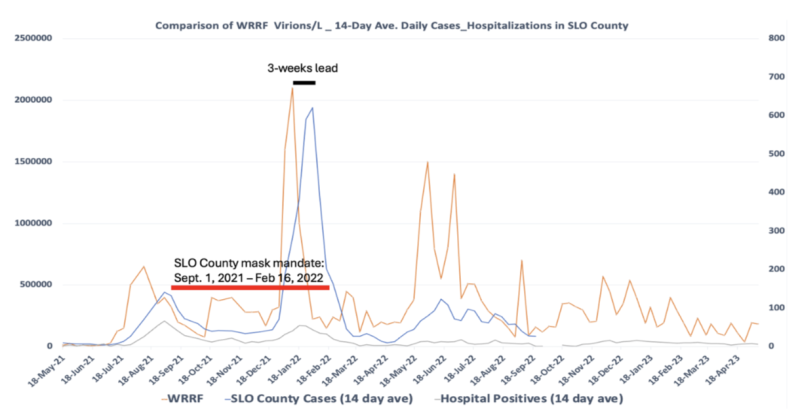
Fig. 7
Taken together, my findings were supported by decades of research showing that masks are not effective against respiratory viruses, regardless of the quality. Still, the counterargument persisted that wearing an N95 mask suctioned to your face, and constantly replacing it, would have stopped the pandemic.
Again, if the cure is not feasible, then it’s not really a cure, is it? The reality is that there are no convincing data supporting mask mandates, none that even remotely support children being forced to wear saliva-soaked masks, and especially none that would justify people being choked and beaten for opposing them.
The “follow the science” crowd was honing their authoritarian skills in preparation for mandatory vaccinations. The motivation for these mandates was summed up perfectly: “During the Sars crisis in 2003 pharma companies answered the WHO’s call for vaccine research. They invested hundreds of millions of dollars, but then — when the outbreak died away — governments and charities lost interest.” According to epidemiologist Dr. Osterholm “The companies were left holding the bag.”
How could Big Pharma avoid “holding the bag” on a vaccine they hoped would stop a virus that had repeatedly ripped through the world’s population? Not surprisingly, their first order of business was to drop the concept of “natural immunity” into the memory hole, centuries of science be damned. The subtext was if regular people knew that natural immunity was real, they probably would not want the vaccine, especially if they already had Covid-19 a few times.
Leading up to the vaccine rollout, I tested myself regularly using PCR, antibody, and antigen assays. I eventually tested positive and had mild flu-like symptoms. While well-educated friends of mine had gone to such lengths as to move out of their homes to distance themselves from their children and wait for the vaccines, my family chose a different tack. Instead, we huddled, got mild infections (except for my wife, who seemed to be immune), shared some level of natural immunity to the latest version of the virus, and tracked our infections (Table 2).
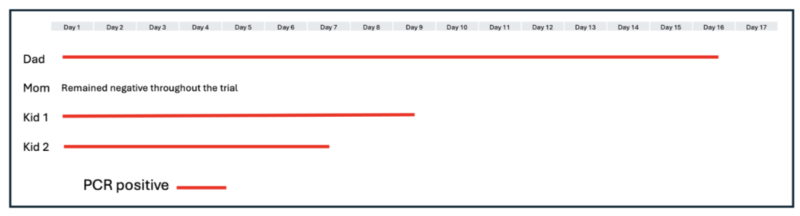
Table 2
When I shared the “herd immunity” story with my small social media following, most appreciated hearing something other than doom and gloom. However, others showed a level of vindictiveness that should not have surprised me, given how acceptable it became to wish death on the unvaccinated.
A colleague attempted to shame me in the campus newspaper, while others wondered out loud whether Child Protective Services should be notified. How dare you give your children the sniffles! How dare you use this time of ridiculous “virtual learning” mandates to provide your children with some hands-on experience performing quantitative PCR!
Predictably, my SARS-CoV-2 antibody levels were extremely high after over two weeks of PCR-positivity. While still overflowing with SARS-CoV-2 antibodies, I was scheduled to receive mandatory shots in order to return to campus.
If the world had actually followed the science, my recent PCR positivity and elevated antibody titers should have been a reasonable exemption. Unfortunately, there was no such exemption. Having seen the terrible treatment of my colleague Dr. Kheriaty, I decided we would play the role of guinea pigs and take what would be an all-risk-and-no-reward shot, especially for my kids. That is, there was nothing in it for us except a few days of high fever and injection site swelling, but definite financial reward for everyone in the vaccine supply chain.
As a member of the “laptop class,” the “lockdowns” made my life easier in many ways. While small business owners struggled, I was getting full pay to upload instructional videos to my university students, and occasionally engage with them online. My wastewater epidemiology work was deemed “essential,” so I was permitted to go to my lab to perform those duties for additional compensation.
However, the ad hominem attacks and threats caused me to disengage from further attempts to start a discussion on pandemic policy, which no doubt was their goal. While the world was fighting over toilet paper and shaming each other for “killing grandma,” we tuned out for a while (Fig. 8).

Fig. 8
I was surrounded by so much anger that I truly believed I was alone in my heretical views on pandemic policy. However, I officially tuned back in when Dr. Scott Atlas invited me to join a small group called The Academy for Science and Freedom.
Our meeting at the Hillsdale College Kirby Center in Washington, D.C. was the first time I had hope since the pandemic started. We were professors, medical doctors, publishers, and journalists, all united by a common belief that the people in charge abandoned a basic tenet of public health: voluntary instead of coercive measures would protect public trust and induce cooperation.
Despite all the great minds in the room, it was hard to imagine we would ever get to where we are right now. But here we are. Many of the people responsible for lockdowns, forced vaccinations, and covering up the unnatural origin of SARS-CoV-2 are gone.
In their place, are Academy members such as Dr. Tracy Beth Høeg, Dr. Jay Bhattacharya, Dr. Matt Memoli, Dr. Vinay Prasad, Dr. Martin Kulldorff, and Dr. Marty Makary. All of whom were treated far worse than me. The overwhelming rejection of “The Fauci School” of public health policy is vindicating. However, recent headlines suggest there are holdouts refusing to accept that they were fooled: Dr. Høeg is a “vaccine skeptic,” Dr. Memoli “is known for questioning vaccine mandates,” and Dr. Prasad is an “anti-science MAHA extremist.”
The people I trusted probably fooled me on a lot of things I voted for, like the benefits of a 20,000-page health care policy. Who has time to actually read that stuff? However, they were never going to succeed at fooling me about the science of the pandemic.
Their lies and hubris caused an awakening, reminiscent of the scene in The Matrix when Neo emerged from the virtual world to a brutal reality. I just hope the people I trust who are now running the major institutions will allocate all resources to programs that will actually improve human health. In doing so, they should have no problem convincing those holdouts not only that they had been fooled, but who fooled them.
New York Times On Climate Change: Two Candidates For Quote Of The Day
By Francis Menton | Manhattan Contrarian | May 21, 2025
Over at the New York Times today, print edition, there is a big front page article documenting how their side is losing the latest battle in the climate wars. The headline is “U.S. Embraces Climate Denial In Science Cuts.” (online headline somewhat different). Also in the Times today (online version) is a feature called “Quote of the Day.” Today’s “quote of the day,” as selected by the Times, is taken from the “climate denial” article just previously linked. Here it is:
“It’s as if we’re in the Dark Ages.”
This quote is attributed to one Rachel Cleetus, identified as senior policy director with the climate and energy program at the Union of Concerned Scientists.
But then, if you take some time to read the article, you come to what I would propose as another excellent candidate for quote of the day. It’s from Brooke Rollins, recently confirmed as the new Secretary of Agriculture in the Trump administration. Here it is:
“We’re not doing that climate change, you know, crud, anymore.”
The focus of the article is what the Times calls “getting rid of data.” In Times spin, the purpose is to “halt the national discussion about how to deal with global warming.” But what kind of “data” are we talking about here? The article is short on specifics as to which exact data series are being cut back or eliminated, let alone whether those series are accurate or useful. But there is enough to give you a general idea:
In recent weeks, more than 500 people have left the National Oceanic and Atmospheric Administration, the government’s premier agency for climate and weather science. . . . NOAA also stopped monthly briefing calls on climate change, and the president’s proposed budget would eliminate funding for the agency’s weather and climate research. The administration has purged the phrases “climate crisis” and “climate science” from government websites.
Ah, NOAA (National Oceanic and Atmospheric Administration). They’re the people who, via their branch called NCEI, put out the so-called “surface temperature” series that have been systematically altered to create a falsely-enhanced warming trend to support regular claims of “warmest day/month/year ever.” This is the subject of my now 33-part series “The Greatest Scientific Fraud Of All Time.”
Let me remind you of the basics of the temperature-alteration scam: (1) the surface temperature records as presented by NOAA/NCEI are not raw instrumental data, but rather have been altered, (2) NOAA admits that it alters the records, (3) NOAA gives seemingly-plausible reasons for altering the records (e.g., to account to station moves and instrument changes), (4) however, the alterations as implemented are not associated with any specific issues like station moves and instrument changes, and (5) the alterations systematically enhance the reported warming trend and are used to support the “climate crisis” narrative. For more detail, go to Part XXXIII of the “Greatest Scientific Fraud” series. Here are just a couple of backup points in case you are skeptical:
-
As to whether NOAA alters the raw data, from ABC News, February 25, 2025, “Yes, NOAA adjusts its historical weather data: Here’s why.” Excerpt: “When digging into conspiracies claiming that the federal agency “manipulates” its historical weather data, ABC News chief meteorologist and chief climate correspondent Ginger Zee was able to confirm that it was true — but that the routine, public adjustments to records happen for good reason. . . . NCEI [a branch of NOAA] adjusts weather data to account for factors like instrument changes, station relocation and urbanization, and it does so through peer-reviewed studies that are published through its federal website.”
-
As to whether the data alterations implemented by NOAA/NCEI can be tied to any specific legitimate bases like station moves or instrumentation changes, I cite a 2022 article by O’Neill, et al. (17 co-authors) from the journal Atmosphere, title “Evaluation of the Homogenization Adjustments Applied to European Temperature Records in the Global Historical Climatology Network Dataset.” I couldn’t get a pithy quote from the article, but here is my summary: “[The authors attempt] to reverse-engineer the adjustments to figure out what NCEI is doing, and particularly whether NCEI is validly identifying station discontinuities, such as moves or instrumentation changes, that might give rise to valid adjustments. The bottom line is that the adjusters make no attempt to tie adjustments to any specific event that would give rise to legitimate homogenization, and that many of the alterations appear ridiculous and completely beyond justification. . . .” There is much, much more detail if you follow the links.
It is not clear from the Times article whether the 500 recent departures from NOAA include the people who have been carrying out this temperature alteration scam. If those people aren’t gone yet, with any luck they will be soon; and maybe we’ll even get some details of how they have been practicing their dark arts.
Meanwhile, back in the world of climate reality, the Real Clear Foundation on Monday (May 19) held something they called the “Energy Future Forum.” Conference co-chairs David DesRosiers and Mark Mills gave opening key-notes. Kevin Killough of Just the News published a summary of the conference on May 20. From DesRosiers’ remarks:
“I think we’ve gone from scarcity to abundance — from the green gospel of scarcity and its Trinitarian ESG god — to the promised land of abundance guided by the values of affordability and reliability,” David DesRosiers, conference co-chair and founder of the RealClear Foundation, said.
And from Mills:
While many tech companies, such as Microsoft, embraced net-zero goals, Mills explained that the energy demands of data centers forced companies to contend with the reality that although fashionable in some circles, intermittent wind and solar power are not adequate. “Eventually, reality rears its ugly head, and we recalibrate around what reality permits,” Mills said.
Bottom line: the Times can scream all it wants, but the world is moving on. From my point of view, it can’t happen too fast.
Top FDA official admits she refused the Covid-19 vaccine while pregnant
A senior regulator’s admission reveals uncomfortable truths about silence, ethics and trust inside the FDA
By Maryanne Demasi, PhD | May 22, 2025
One of the most powerful figures at the US Food and Drug Administration (FDA) has admitted she refused the Covid-19 mRNA vaccine while pregnant—even as her agency promoted it as “safe and effective” for all pregnant women.
Dr Sara Brenner’s explosive disclosure, made on 15 May 2025 at the MAHA Institute Round Table in Washington DC, is as revealing as it is troubling.
A preventive medicine physician, Brenner has worked at the FDA since 2019. As the FDA’s Principal Deputy Commissioner—and briefly its Acting Commissioner—Brenner was at the centre of decision-making.

Dr Sara Brenner on 15 May 2025 at the MAHA Institute
Prior to that she was Chief Medical Officer for diagnostics and was detailed to the White House to support the Biden administration’s Covid-19 response. She didn’t just participate in the pandemic response, she helped shape it from within.
“Knowing what I knew—not only about nanotechnology, about medicine, about the medical countermeasures—but also having a very strong and firm grounding in bioethics… there were many things that were not right,” she told the audience.
That someone with her seniority and access to internal data privately rejected the vaccine, while her agency promoted it to millions of pregnant women, presents a profound ethical dilemma.
Brenner’s concerns about mRNA safety
Brenner explained that her decision was driven by a lack of safety data, particularly around the biodistribution of the vaccine’s lipid nanoparticles (LNPs)—the tiny fat particles used to deliver the mRNA into cells.
“It was unknown at the time what the biodistribution patterns of those products were… That was my primary concern, and that exposure I was very concerned about,” said Brenner.
She had reason to be cautious.
As a nanomedicine expert who built an MD/PhD program in the field, Brenner had spent years researching the “biodistribution, excretion, metabolism and toxicities associated with engineered nanoparticles.”
“Materials that don’t exist in nature—there’s a lot of unknowns,” said Brenner.
She warned that unintended toxic effects—especially in vulnerable populations like pregnant women—could not be ignored.
“Regardless of the medical product or the intervention, there’s always going to be the need to evaluate both the intended outcomes… and the unintended consequences,” she cautioned.
Warnings ignored
Brenner’s concerns echoed those raised in 2021 by Canadian immunologist Dr Byram Bridle, who first exposed internal documents from Japan’s regulatory agency showing that LNPs didn’t remain at the injection site, but travelled throughout the body and accumulated in organs including the ovaries, liver, spleen and bone marrow.
At the time, Bridle’s warnings were aggressively dismissed. His reputation took a hit, and he faced institutional censure from the University of Guelph, where he was a professor, for speaking out against vaccine mandates.

Dr Byram Bridle, Canadian immunologist. Photo credit: Kenneth Armstrong
Now, Brenner’s comments confirm that these concerns were not only valid—they were quietly shared at the highest levels of the FDA.
During the event, Brenner also revealed that her worries extended to breastfeeding and potential exposure to her child after birth.
A 2022 study published in JAMA Pediatrics detected vaccine-derived mRNA in the breast milk of vaccinated mothers for at least 48 hours—the very scenario Brenner had feared.
Yet the FDA made little effort to publicly investigate or address the findings, dismissing them with the vague reassurance that there was “no evidence of harm.”
No mandate for Brenner?
It’s unclear how Brenner managed to avoid the vaccine mandate that applied to all federal employees at the time. She didn’t say. Perhaps she received a religious or medical exemption—but she left that part out.
What she did reveal was that she had concerns—deep enough not to take the vaccine during her pregnancy. Yet she said nothing publicly, while her agency told millions of other women it was safe.
For many, that silence is hard to accept and it has left many asking why she didn’t warn other women about a product with ‘zero’ clinical safety data in pregnancy.
No one but Brenner knows the full story. But the ethical contradiction is hard to ignore.
Silence inside the castle
Brenner acknowledged the immense pressure inside the FDA to stick to the official narrative.
“They don’t let you get very far out of the castle at FDA with your talking points,” she admitted nervously.
She described the period as a “dark night of the soul” for many civil servants, a time when even “very obvious things” took bravery to say.
She eventually found support through a group called Feds for Medical Freedom—federal workers advocating for informed consent, bodily autonomy, and pushing back against government overreach.
A culture change?
Today, under a new administration, Brenner says the culture inside the FDA is shifting. She praised Commissioner Dr Marty Makary and said transparency is finally becoming a priority.
“We’re moving very quickly to make it such that there will be more transparency… so that people can see and evaluate for themselves what the truths are.”
But Brenner’s remarks won’t undo what has already happened—especially to those who were vaccine injured or whose pregnancies were affected.
What her comments do offer is a rare glimpse into the internal dynamics of a government institution that issued sweeping public assurances while failing to acknowledge its own uncertainty.
“There was no acknowledgement of what was unknown. There were only statements and assertions that were really more like beliefs,” Brenner said of the FDA’s messaging during the pandemic.
That may be her most important admission.
This is more than a story about one woman’s personal decision. It is a story about institutional culture, regulatory failure, and the consequences of silence.
Those who spoke up were punished. Those who stayed silent kept their jobs and reputations. And those who were forced to comply were often left to deal with the collateral damage.
When asked whether she believed she had made the right decision in refusing the Covid-19 vaccine, Brenner replied simply, “I believe so.”
Now that she has spoken, the question remains — who else knew, and said nothing?
Featured Video

US Middle East Policy: The Growing Propensity for Genocide
or go to
Aletho News Archives – Video-Images
From the Archives
The New Baghdad Pact
By Dr Bouthaina Shaaban | February 23, 2017
A recently declassified CIA document prepared in 1983, and released on 20 January 2017, shows that the United States had at the time encouraged Saddam Hussein to attack Syria, which would have led to a vicious conflict between the two countries, thus draining their resources.
The report, which was then prepared by CIA officer Graham Fuller, indicates that the US tried adamantly to convince Saddam to attack Syria under any pretense available, in order to get the two most powerful countries in the Arab East to destroy each other, turning their attention away from the Arab-Israeli conflict. … continue
Blog Roll
 Aletho News
Aletho News- The Gratitude of the Captured
- Hezbollah denies involvement in deadly attack on UNIFIL in south Lebanon
- The prospect of an expanded and far more violent war
- Canada’s Carney Revives Online Censorship Bill
- Israeli soldiers kill UNICEF truck drivers delivering water to Gaza families
- Iran defends limits on Strait of Hormuz passage
- ‘We warned you’: Hormuz Strait back to pervious state amid US blockade
- Iran rejects uranium transfer, warns of response to naval blockade
- US Middle East Policy: The Growing Propensity for Genocide
- Daniel Davis: Iran Reopens the Strait of Hormuz
- If Americans Knew
- Israel relegates another population to life in tents – Daily Update
- Senate again fails to block weapons to Israel
- Think the Iran war is a disaster? Blame these DC think tanks first.
- Number of Palestinian Prisoners Rises By 83% Since October 2023
- With multiple “ceasefires” in place, Israel keeps on killing in Gaza and Lebanon – Daily Update
- Mearsheimer: Israel Owns Trump
- Mark Levin and Jonathan Pollard Push for Nuking Iran
- TCN: America Enables Israel’s Crimes
- Israel’s Next Leader Will Be Just Like Bibi – but Without the Corruption
- Born into war, raised across borders: The story of Gaza’s premature babies separated from their parents amid Israel’s genocide
- No Tricks Zone
- Reality Check: Maldives Have Actually Grown In Size Or Remained Stable Over Recent Decades
- Abrupt Climate Change Also Occurred NATURALLY In The Past …25 Times During Last Ice Age
- Cave Discovery Reveals Today’s Desert Climates Were Recently Far Warmer, Wetter, Teeming With Life
- German Expert: Heat Dome Led To Record Temps In Western USA…Warmer In 1934, 1936
- New Study: No Linear Warming Or Glacier Retreat Along Northern Antarctic Peninsula Since 1980s
- An Inconvenient Tree: Uncovered In Alps… Europe Much Warmer Than Today 6000 Years Ago
- New Study Reports A 60% Slowdown In Greenland’s Ice Loss Rate In The Last Decade
- Low Intensity Tornado Wrecks Major Solar Farm, Creating A Potential Toxic Dump
- New Study Finds Warming Saves Lives…Cold Temperatures 12 Times More Deadly Than Excess Heat
- German Science Blog Accuses PIK Climate Institute Of Hallucinating Climate Tipping Points
