APOE-4: The Clue to Why Low Fat Diet and Statins may Cause Alzheimer’s
By Dr. Stephanie Seneff | December 15, 2009
Abstract
Alzheimer’s is a devastating disease whose incidence is clearly on the rise in America. Fortunately, a significant number of research dollars are currently being spent to try to understand what causes Alzheimer’s. ApoE-4, a particular allele of the apolipoprotein apoE, is a known risk factor. Since apoE plays a critical role in the transport of cholesterol and fats to the brain, it can be hypothesized that insufficient fat and cholesterol in the brain play a critical role in the disease process. In a remarkable recent study, it was found that Alzheimer’s patients have only 1/6 of the concentration of free fatty acids in the cerebrospinal fluid compared to individuals without Alzheimer’s. In parallel, it is becoming very clear that cholesterol is pervasive in the brain, and that it plays a critical role both in nerve transport in the synapse and in maintaining the health of the myelin sheath coating nerve fibers. An extremely high-fat (ketogenic) diet has been found to improve cognitive ability in Alzheimer’s patients. These and other observations described below lead me to conclude that both a low-fat diet and statin drug treatment increase susceptibility to Alzheimer’s.
1. Introduction
Alzheimer’s is a devastating disease that takes away the mind bit by bit over a period of decades. It begins as odd memory gaps but then steadily erodes your life to the point where around-the-clock care is the only option. With severe Alzheimer’s, you can easily wander off and get lost, and may not even recognize your own daughter. Alzheimer’s was a little known disease before 1960, but today it threatens to completely derail the health system in the United States.
Currently, over 5 million people in America have Alzheimer’s. On average, a person over 65 with Alzheimer’s costs three times as much for health care as one without Alzheimer’s. More alarmingly, the incidence of Alzheimer’s is on the rise. Dr. Murray Waldman has studied epidemiological data comparing Alzheimer’s with femur fractures, looking back over the last fifty years [52]. Alarmingly, he has found that, while the incidence of femur fractures (another condition which typically increases with age) has gone up only at a linear rate, the increase in the incidence of Alzheimer’s has gone up exponentially, between 1960 and 2010 Alzheimer’s Epidemic [15]. Just between 2000 and 2006, US Alzheimer’s deaths rose by 47%, while, by comparison, deaths from heart disease, breast cancer, prostate cancer, and stroke combined decreased by 11%. This increase goes far beyond people living longer: for people 85 and older, the percentage who died from Alzheimer’s rose by 30% between 2000 and 2005 [2]. Finally, it’s likely these are under-estimates, as many people suffering with Alzheimer’s ultimately die of something else. You likely have a close friend or relative who is suffering from Alzheimer’s.
Something in our current lifestyle is increasing the likelihood that we will succumb to Alzheimer’s. My belief is that two major contributors are our current obsession with low-fat diet, combined with the ever expanding use of statin drugs. I have argued elsewhere that low-fat diet may be a major factor in the alarming increase in autism and adhd in children. I have also argued that the obesity epidemic and the associated metabolic syndrome can be traced to excessive low-fat diet. Statins are likely contributing to an increase in many serious health issues besides Alzheimer’s, such as sepsis, heart failure, fetal damage, and cancer, as I have argued here. I believe the trends will only get worse in the future, unless we substantially alter our current view of “healthy living.”
The ideas developed in this essay are the result of extensive on-line research I conducted to try to understand the process by which Alzheimer’s develops. Fortunately, a great deal of research money is currently being spent on Alzheimer’s, but a clearly articulated cause is still elusive. However, many exciting leads are fresh off the press, and the puzzle pieces are beginning to assemble themselves into a coherent story. Researchers are only recently discovering that both fat and cholesterol are severly deficient in the Alzheimer’s brain. It turns out that fat and cholesterol are both vital nutrients in the brain. The brain contains only 2% of the body’s mass, but 25% of the total cholesterol. Cholesterol is essential both in transmitting nerve signals and in fighting off infections.
A crucial piece of the puzzle is a genetic marker that predisposes people to Alzheimer’s, termed “apoE-4.” ApoE plays a central role in the transport of fats and cholesterol. There are currently five known distinct variants of apoE (properly termed “alleles”), with the ones labelled “2”, “3” and “4” being the most prevalent. ApoE-2 has been shown to afford some protection against Alzheimer’s; apoE-3 is the most common “default” allele, and apoE-4, present in 13-15% of the population, is the allele that is associated with increased risk to Alzheimer’s. A person with apoE-4 allele inherited from both their mother and their father has up to a twenty-fold increased likelihood of developing Alzheimer’s disease. However, only about 5% of the people with Alzheimer’s actually have the apoE-4 allele, so clearly there is something else going on for the rest of them. Nonetheless, understanding apoE’s many roles in the body was a key step leading to my proposed low fat/statin theory.
2. Background: Brain Biology 101
Although I have tried to write this essay in a way that is accessible to the non-expert, it will still be helpful to first familiarize you with basic knowledge of the structure of the brain and the roles played by different cell types within the brain.
At the simplest level, the brain can be characterized as consisting of two major components: the gray matter and the white matter. The gray matter comprises the bodies of the neurons, including the cell nucleus, and the white matter contains the myriad of “wires” that connect each neuron to every other neuron it communicates with. The wires are known as “axons” and they can be quite long, connecting, for example, neurons in the frontal cortex (above the eyes) with other neurons deep in the interior of the brain concerned with memory and movement. The axons will figure prominently in the discussions below, because they are coated with a fatty substance called the myelin sheath, and this insulating layer is known to be defective in Alzheimer’s. Neurons pick up signals transmitted through the axons at junctures known as synapses. Here the message needs to be transmitted from one neuron to another one, and various neurotransmitters such as dopamine and GABA exert excitatory or inhibitory influences on signal strength. In adidtion to a single axon, neurons typically have several much shorter nerve fibers called dendrites, whose job is to receive incoming signals from diverse sources. At a given point in time, signals received from multiple sources are integrated in the cell body and a decision is made as to whether the accumulated signal strength is above threshold, in which case the neuron responds by firing a sequence of electrical pulses, which are then transmitted through the axon to a possibly distant destination.
In addition to the neurons, the brain also contains a large number of “helper” cells called glial cells, which are concerned with the care and feeding of neurons. Three principle types of glial cells will play a role in our later discussion: the microglia, the astrocytes, and the oligodendrocytes. Microglia are the equivalent of white blood cells in the rest of the body. They are concerned with fighting off infective agents such as bacteria and viruses, and they also monitor neuron health, making life-and-death decisions: programming a particular neuron for apoptosis (intentional self-destruction) if it appears to be malfunctioning beyond hope of recovery, or is infected with an organism that is too dangerous to let flourish.
The astrocytes figure very prominently in our story below. They nestle up against the neurons and are responsible for assuring an adequate supply of nutrients. Studies on neuron cultures from rodent central nervous systems have shown that neurons depend upon astrocytes for their supply of cholesterol [40]. Neurons critically need cholesterol, both in the synapse [50] and in the myelin sheath [45], in order to successfully transmit their signals, and also as a first line of defense against invasive microbes. Cholesterol is so important to the brain that astrocytes are able to synthesize it from basic ingredients, a skill not found in most cell types. They also supply the neurons with fatty acids, and they are able to take in short chain fatty acids and combine them to form the longer-chain types of fatty acids that are especially prominent in the brain [7][24][36], and then deliver them to neighboring neurons and to the cerebrospinal fluid.
The third type of glial cell is the oligodendrocyte. These cells specialize in making sure the myelin sheath is healthy. Oligodentrocytes synthesize a special sulfur-containing fatty acid, known as sulfatide, from other fatty acids supplied to them by the cerebrospinal fluid [9]. Sulfatide has been shown to be essential for the maintenance of the myelin sheath. Children born with a defect in the ability to metabolize sulfatide suffer from progressive demyelination, and rapid loss of motor and cognitive functions, resulting in an early death before the age of 5 [29]. Depletion in sulfatide is a well-known characterization of Alzheimer’s, even in early stages before it has been manifested as cognitive decline [18]. And ApoE has been shown to play a crucial role in the maintenance of sulfatide [19]. Throughout a person’s life, the myelin sheath has to be constantly maintained and repaired. This is something that researchers are only beginning to appreciate, but two related properties of Alzheimer’s are poor quality myelin sheath alongside a drastically reduced concentration of fatty acids and cholesterol in the cerebrospinal fluid [38].
3. Cholesterol and Lipid Management
In addition to some knowledge about the brain, you will also need to know something about the processes that deliver fats and cholesterol to all the tissues of the body, with a special focus on the brain. Most cell types can use either fats or glucose (a simple sugar derived from carbohydrates) as a fuel source to satisfy their energy needs. However, the brain is the one huge exception to this rule. All cells in the brain, both the neurons and the glial cells, are unable to utilize fats for fuel. This is likely because fats are too precious to the brain. The myelin sheath requires a constant supply of high quality fat to insulate and protect the enclosed axons. Since the brain needs its fats to survive long-term, it is paramount to protect them from oxidation (by exposure to oxygen) and from attack by invasive microbes.
Fats come in all kinds of shapes and sizes. One dimension is the degree of saturation, which concerns how many double bonds they possess, with saturated fats possessing none, monounsaturated fats having only one, and polyunsaturated fats having two or more. Oxygen breaks the double bond and leaves the fat oxidized, which is problematic for the brain. Polyunsaturated fats are thus the most vulnerable to oxygen exposure, because of multiple double bonds.
Fats are digested in the intestine and released into the blood stream in the form of a relatively large ball with a protective protein coat, called a chylomicron. The chylomicron can directly provide fuel to many cell types, but it may also be sent to the liver where the contained fats are sorted out and redistributed into much smaller particles, which also contain substantial amounts of cholesterol. These particles are called “lipoproteins,” (henceforth, LPP’s) because they contain protein in the spherical shell and lipids (fats) in the interior. If you’ve had your cholesterol measured, you’ve probably heard of LDL (low density LPP) and HDL (high density LPP). If you think these are two different kinds of cholesterol, you would be mistaken. They are just two different kinds of containers for cholesterol and fats that serve different roles in the body. There are actually several other LPP’s, for example, VLDL (very-low) and IDL (intermediate), as shown in the accompanying diagram. 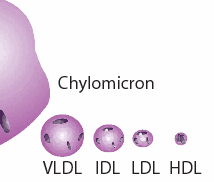 In this essay I will refer to these collectively as the XDL’s. As if this weren’t confusing enough, there is also another unique XDL that is found only in the cerebrospinal fluid, that supplies the nutritional needs of the brain and nervous system. This one doesn’t seem to have a name yet, but I will call it “B-HDL,” because it is like HDL in terms of its size, and “B” is for “brain [13]”
In this essay I will refer to these collectively as the XDL’s. As if this weren’t confusing enough, there is also another unique XDL that is found only in the cerebrospinal fluid, that supplies the nutritional needs of the brain and nervous system. This one doesn’t seem to have a name yet, but I will call it “B-HDL,” because it is like HDL in terms of its size, and “B” is for “brain [13]”
An important point about all the XDL’s is that they contain distinctly different compositions, and each is targeted (programmed) for specific tissues. A set of proteins called “apolipoproteins” or, equivalently, “apoproteins” (“apo’s” for short) figure strongly in controlling who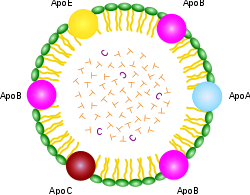 gets what. As you can see from the schematic of the chylomicron shown at the right, it contains a rainbow of different apo’s for every conceivable application. But the XDL’s are far more specific, with HDL containing “A,” LDL containing “B,” VLDL containing “B” and “C,” and IDL containing only “E.” The apo’s have special binding properties that allow the lipid contents to be transported across cell membranes so that the cell can gain access to the fats and choleseterol contained inside.
gets what. As you can see from the schematic of the chylomicron shown at the right, it contains a rainbow of different apo’s for every conceivable application. But the XDL’s are far more specific, with HDL containing “A,” LDL containing “B,” VLDL containing “B” and “C,” and IDL containing only “E.” The apo’s have special binding properties that allow the lipid contents to be transported across cell membranes so that the cell can gain access to the fats and choleseterol contained inside.
The only apo that is of concern to us in the context of this essay is apoE. ApoE is very important to our story because of its known link with Alzheimer’s disease. ApoE is a protein, i.e., sequence of amino acids, and its specific composition is dictated by a corresponding DNA sequence on a protein-coding gene. Certain alterations in the DNA code lead to defects in the ability of the transcribed protein to perform its biological roles. ApoE-4, the allele associated with increased risk to Alzheimer’s, is presumably unable to perform its tasks as efficiently as the other alleles. By understanding what apoE does, we can better infer how the consequences of doing it poorly might impact the brain, and then observe experimentally whether the features of the Alzheimer’s brain are consistent with the roles played by apoE.
A strong clue about apoE’s roles can be deduced from where it is found. As I mentioned above, it is the only apo in both B-HDL in the cerebrospinal fluid and IDL in the blood serum. Only selected cell types can synthesize it, the two most significant of which for our purposes are the liver and the astrocytes in the brain. Thus the astrocytes provide the linkage between the blood and the cerebrospinal fluid. They can usher lipids and cholesterol across the blood-brain barrier, via the special key which is apoE.
It turns out that, although apoE is not found in LDL, it does bind to LDL, and this means that astrocytes can unlock the key to LDL in the same way that they can gain access to IDL, and hence the cholesterol and fatty acid contents of LDL are accessible to astrocytes as well, as long as apoE is functioning properly. The astrocytes reshape and repackage the lipids and release them into the cerebospinal fluid, both as B-HDL and simply as free fatty acids, available for uptake by all parts of the brain and nervous system [13].
One of the critical reshaping steps is to convert the fats into types that are more attractive to the brain. To understand this process you need to know about another dimension of fats besides their degree of saturation, which is their total length. Fats have a chain of linked carbon atoms as their spine, and the total number of carbons in a particular fat characterizes it as short, medium length, or long. The brain works best when the constituent fats are long, and, indeed, the astrocytes are able to take in short chain fats and reorganize them to make longer chain fats [24].
A final dimension of fats that plays a role is where the first double bond is located in a polyunsaturated fat, which distinguishes omega-3 from omega-6 fats (position 3; position 6). Omega-3 fats are very common in the brain. Certain ones of the omega-3 and omega-6 fats are essential fatty acids, in that the human body is unable to synthesize them, and therefore depends upon their supply from the diet. This is why it is claimed that fish “makes you smart”: because cold water fish is the best source of essential omega-3 fats.
Now I want to return to the subject of the XDL’s. It is a dangerous journey from the liver to the brain, as both oxygen and microbes are found in abundance in the blood stream. The XDL’s protective shell contains both LPP’s andunesterified cholesterol, as well as the signature apo that controls which cells can receive the contents, as shown in the accompanying schematic. 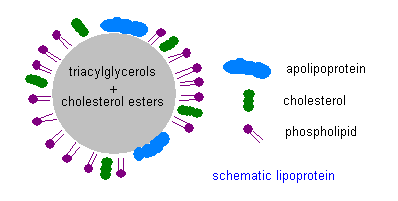 The internal contents are esterified cholesterol and fatty acids, along with certain antioxidants that are conveniently being transported to the cells packaged in the same cargo ship. Esterification is a technique to render the fats and cholesterol inert, which helps protect them from oxidation [51]. Having the antioxidants (such as vitamin E and Coenzyme Q10) along for the ride is also convenient, as they too protect against oxidation. The cholesterol contained in the shell, however, is intentionally not esterified, which means that it is active. One of its roles there is to guard against invasive bacteria and viruses [55]. Cholesterol is the first line of defense against these microbes, as it will alert the white blood cells to attack whenever it encounters dangerous pathogens. It has also been proposed that the cholesterol in the XDL’s shell itself acts as an antioxidant [48].
The internal contents are esterified cholesterol and fatty acids, along with certain antioxidants that are conveniently being transported to the cells packaged in the same cargo ship. Esterification is a technique to render the fats and cholesterol inert, which helps protect them from oxidation [51]. Having the antioxidants (such as vitamin E and Coenzyme Q10) along for the ride is also convenient, as they too protect against oxidation. The cholesterol contained in the shell, however, is intentionally not esterified, which means that it is active. One of its roles there is to guard against invasive bacteria and viruses [55]. Cholesterol is the first line of defense against these microbes, as it will alert the white blood cells to attack whenever it encounters dangerous pathogens. It has also been proposed that the cholesterol in the XDL’s shell itself acts as an antioxidant [48].
HDL’s are mostly depleted of the lipid and cholesterol content, and they are tasked with returning the empty shell back to the liver. Once there, cholesterol will be recommissioned to enter the digestive system as part of the bile, which is produced by the gall bladder to help digest ingested fats. But the body is very careful to conserve cholesterol, so that 90% of it will be recycled from the gut back into the blood stream, contained in the chylomicron that began our story about fats.
In summary, the management of the distribution of fats and cholesterol to the cells of the body is a complex process, carefully orchestrated to assure that they will have a safe journey to their destination. Dangers lurk in the blood stream, mostly in the form of oxygen and invasive microbes. The body considers cholesterol to be precious cargo, and it is very careful to conserve it, by recycling it from the gut back to the liver, to be appropriately distributed among the XDL’s that will deliver both cholesterol and fats to the tissues that depend upon them, most especially the brain and nervous system.
4. The Relationship between Cholesterol and Alzheimer’s
Through retrospective studies, the statin industry has been very successful at the game of pretending that benefits derived from high cholesterol are actually due to statins, as I have described at length in an essay on the relationship between statins and fetal damage, sepsis, cancer, and heart failure. In the case of Alzheimer’s, they are playing this game in reverse: they are blaming cholesterol for a very serious problem that I believe is actually caused by statins.
The statin industry has looked long and hard for evidence that high cholesterol might be a risk factor for Alzheimer’s. They examined cholesterol levels for men and women of all ages between 50 and 100, looking back 30 or more years if necesssary, to see if there was ever a correlation between high cholesterol and Alzheimer’s. They found only one statistically significant relationship: men who had had high cholesterol in their 50’s had an increased susceptibility to Alzheimer’s much later in life [3].
The statin industry has jumped on this opportunity to imply that high cholesterol might cause Alzheimer’s, and, indeed, they have been very fortunate in that reporters have taken the bait and are promoting the idea that, if high cholesterol many years ago is linked to Alzheimer’s, then statins might protect from Alzheimer’s. Fortunately, there exist lengthy web pages (Cholesterol Doesn’t Cause Alzheimer’s) that have documented the long list of reasons why this idea is absurd.
Men who have high cholesterol in their 50’s are the poster child for statin treatment: all of the studies that have shown a benefit for statins in terms of reducing the number of minor heart attacks involved men in their 50’s. High cholesterol is positively correlated with longevity in people over 85 years old [54], and has been shown to be associated with better memory function [53] and reduced dementia [35]. The converse is also true: a correlation between falling cholesterol levels and Alzheimer’s [39]. As will be discussed further later, people with Alzheimer’s also have reduced levels of B-HDL, as well as sharply reduced levels of fatty acids, in the cerbrospinal fluid, i.e, impoverished supply of cholesterol and fats to the myelin sheath [38]. As we saw earlier, fatty acid supply is essential as building blocks for the sulfatide that is synthesized by oligodendrocytes to keep the myelin sheath healthy [29].
The obvious study that needs to be done is to bin the men who had high cholesterol in their 50’s into three groups: those who never took statins, those who took smaller doses for shorter times, and those who took larger doses for longer times. Such a study would not be hard to do; in fact, I suspect something like it has already been done. But you’ll never hear about it because the statin industry has buried the results.
In a very long term retrospective cohort study of members of the Permanente Medical Care Program in northern California, researchers looked at cholesterol data that were obtained between 1964 and 1973 [46]. They studied nearly ten thousand people who had remained members of that health plan in 1994, upon the release of computerized outpatient diagnoses of dementia (both Alzheimer’s and vascular dementia). The subjects were between 40 and 45 years old when the cholesterol data were collected.
The researchers found a barely statistically significant result that people who were diagnosed with Alzheimer’s had higher cholesterol in their 50’s than the control group. The mean value for the Alzheimer’s patients was 228.5, as against 224.1 for the controls.
The question that everybody ought to be asking is: for the Alzheimer’s group, how did the people who later took statins stack up against the people who didn’t? In extreme understatement, the authors offhandedly remark in the middle of a paragraph: “Information on lipid-lowering treatments, which have been suggested to decrease dementia risk [31], was not available for this study.” You can be sure that, if there was any inkling that the statins might have helped, these researchers would have been allowed access to those data.
The article they refer to for support, reference [19] in [46] (which is reference [44] here) was very weak. The abstract for that article is repeated in full here in the Appendix. But the concluding sentence sums it up well: “A more than a modest role for statins in preventing AD [Alzheimer’s Disease] seems unlikely.” This is the best they can come up with to defend the position that statins might protect from Alzheimer’s.
An intuitive explanation for why high cholesterol at an early age might be correlated with Alzheimer’s risk has to do with apoE-4. People with that allele are known to have high cholesterol early in life [39], and I believe this is a protective strategy on the part of the body. The apoE-4 allele is likely defective in the task of importing cholesterol into the astrocytes, and therefore an increase in the bioavailability of cholesterol in blood serum would help to offset this deficit. Taking a statin would be the last thing a person in that situation would want to do.
5. Do Statins Cause Alzheimer’s?
There is a clear reason why statins would promote Alzheimer’s. They cripple the liver’s ability to synthesize cholesterol, and as a consequence the level of LDL in the blood plummets. Cholesterol plays a crucial role in the brain, both in terms of enabling signal transport across the synapse [50] and in terms of encouraging the growth of neurons through healthy development of the myelin sheath [45]. Nonetheless, the statin industry proudly boasts that statins are effective at interfering with cholesterol production in the brain [31][47] as well as in the liver.
Yeon-Kyun Shin is an expert on the physical mechanism of cholesterol in the synapse to promote transmission of neural messages, and one of the authors of [50] referenced earlier. In an interview by a Science Daily reporter, Shin said: “If you deprive cholesterol from the brain, then you directly affect the machinery that triggers the release of neurotransmitters. Neurotransmitters affect the data-processing and memory functions. In other words — how smart you are and how well you remember things.”
A recent review of two large population-based double-blind placebo-controlled studies of statin medications in individuals at risk for dementia and Alzheimer disease showed that statins are not protective against Alzheimer’s [34]. The lead author of the study, Bernadette McGuinness, was quoted by a reporter from Science Daily as saying, “From these trials, which contained very large numbers and were the gold standard — it appears that statins given in late life to individuals at risk of vascular disease do not prevent against dementia.” A researcher at UCLA, Beatrice Golomb, when asked to comment on the results, was even more negative, saying, “Regarding statins as preventive medicines, there are a number of individual cases in case reports and case series where cognition is clearly and reproducibly adversely affected by statins.” In the interview, Golomb remarked that various randomized trials have shown that statins were either adverse or neutral towards cognition, but none have shown a favorable response.
A common side effect of statins is memory dysfunction. Dr. Duane Graveline, fondly known as “spacedoc” because he served as a doctor to the astronauts, has been a strong advocate against statins and is collecting evidence of statin side effects directly from statin users around the world. He was led to this assault on statins as a consequence of his own personal experience of transient global amnesia, a frightening episode of total memory loss which he is convinced was caused by the statin drugs he was taking at the time. He has now completed three books describing a diverse collection of damning side effects of statins, the most famous of which is Lipitor: Thief of Memory [17].
A second way (besides their direct impact on cholesterol) in which statins likely impact Alzheimer’s is in their indirect negative effect on the supply of fatty acids and antioxidants to the brain. It is a given that statins drastically reduce the level of LDL in the blood serum. This is their claim to fame. It is interesting, however, that they succeed in reducing not just the amount of cholesterol contained in the LDL particles, but rather the actual number of LDL particles altogether. This means that, in addition to depleting cholesterol, they reduce the available supply to the brain of both fatty acids and antixodiants, which are also carried in the LDL particles. As we’ve seen, all three of these substances are essential to proper brain functioning.
I conjecture that the reasons for this indirect effect are two-fold: (1) there is inadequate cholesterol in the bile to metabolize dietary fats, and (2) the rate-limiting effect on the production of LDL is the ability to provide adequate cholesterol in the shell to assure survival of the contents during transport in the blood stream; i.e., to protect the contents from oxidation and marauding bacteria and viruses. People who take the highest 80 mg/dl dosage of statins often end up with LDL levels as low as 40mg/dl, well below even the lowest numbers observed naturally. I shudder to think of the probable long-term consequences of such severe depletion in fats, cholesterol, and antioxidants.
A third way in which statins may promote Alzheimer’s is by crippling the ability for cells to synthesize coenzyme Q10. Coenzyme Q10 has the misfortune of sharing the same metabolic pathway as cholesterol. Statins interfere with a crucial intermediate step on the pathway to the synthesis of both cholesterol and coenzyme Q10. Coenzyme Q10 is also known as “ubiquinone” because it seems to show up everywhere in cell metabolism. It is found both in the mitochondria and in the lysosomes, and its critical role in both places is as an antioxidant. The inert esters of both cholesterol and fatty acids are hydrolyzed and activated in the lysosomes [8], and then released into the cytoplasm. Coenzyme Q10 consumes excess oxygen to keep it from doing oxidative damage [30], while also generating energy in the form of ATP (adenosine triphosphate, the universal energy currency in biology).
The final way in which statins should increase Alzheimer’s risk is through their indirect effect on vitamin D. 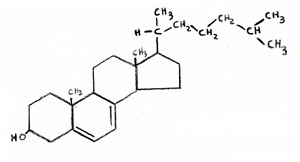 Vitamin D is synthesized from cholesterol in the skin, upon exposure to UV rays from the sun. In fact, the chemical formula of vitamin D is almost indistinguishable from that of cholesterol, as shown in the two attached figures (cholesterol on the left, vitamin D on the right). If LDL levels are
Vitamin D is synthesized from cholesterol in the skin, upon exposure to UV rays from the sun. In fact, the chemical formula of vitamin D is almost indistinguishable from that of cholesterol, as shown in the two attached figures (cholesterol on the left, vitamin D on the right). If LDL levels are 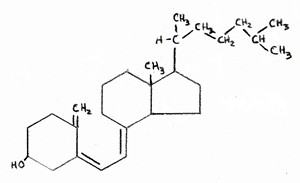 kept artificially low, then the body will be unable to resupply adequate amounts of cholesterol to replenish the stores in the skin once they have been depleted. This would lead to vitamin D deficiency, which is a widespread problem in America.
kept artificially low, then the body will be unable to resupply adequate amounts of cholesterol to replenish the stores in the skin once they have been depleted. This would lead to vitamin D deficiency, which is a widespread problem in America.
It is well known that vitamin D fights infection. To quote from [25], “Patients with severe infections as in sepsis have a high prevalence of vitamin D deficiency and high mortality rates.” As will be elaborated on later, a large number of infective agents have been shown to be present in abnormally high amounts in the brains of Alzheimers patients [27][26].
Dr. Grant has recently argued [16] that there are many lines of evidence pointing to the idea that dementia is associated with vitamin D deficiency. An indirect argument is that vitamin D deficiency is associated with many conditions that in turn carry increased risk for dementia, such as diabetes, depression, osteoporosis, and cardiovascular disease. Vitamin D receptors are widespread in the brain, and it is likely that they play a role there in fighting off infection. Vitamin D surely plays other vital roles in the brain as well, as powerfully suggested by this quote taken from the abstract of [32]: “We conclude there is ample biological evidence to suggest an important role for vitamin D in brain development and function.”
6. Astrocytes, Glucose Metabolism, and Oxygen
Alzheimer’s is clearly correlated with a deficiency in the supply of fat and cholesterol to the brain. IDL, when functioning properly, is actually incredibly efficient in cholesterol and fat throughput from the blood across cell membranes, compared to LDL [8]. It gives up its contents much more readily than the other apo’s. And it achieves this as a direct consequence of apoE. IDL (as well as LDL) in the blood delivers fats and cholesterol to the astrocytes in the brain, and the astrocytes can thus use this external source instead of having to produce these nutrients themselves. I suspect, in fact, that astrocytes only produce a private supply when the external supply is insufficient, and they do so reluctantly.
Why would it be disadvantageous for an astrocyte to synthesize its own fats and cholesterol? In my opinion, the answer has to do with oxygen. An astrocyte needs a significant energy source to synthesize fats and cholesterol, and this energy is usually supplied by glucose from the blood stream. Furthermore, the end-product of glucose metabolism is acetyl-Coenzyme A, the precursor to both fatty acids and cholesterol. Glucose can be consumed very efficiently in the mitochondria, internal structures within the cell cytoplasm, via aerobic processes that require oxygen. The glucose is broken down to produce acetyl-Coenzyme A as an end-product, as well as ATP, the source of energy in all cells.
However, oxygen is toxic to lipids (fats), because it oxidizes them and makes them rancid. Lipids are fragile if not encased in a protective shell like IDL, HDL, or LDL. Once they are rancid they are susceptible to infection by invasive agents like bacteria and viruses. So an astrocyte trying to synthesize a lipid has to be very careful to keep oxygen out, yet oxygen is needed for efficient metabolism of glucose, which will provide both the fuel (ATP) and the raw materials (acetyl-Coenzyme A) for fat and cholesterol synthesis.
What to do? Well, it turns out that there is an alternative, although much less efficient, solution: to metabolize glucose anaerobically directly in the cytoplasm. This process does not depend on oxygen (a great advantage) but it also yields substantially less ATP (only 6 ATP as contrasted with 30 if glucose is metabolized aerobically in the mitochondria). The end product of this anaerobic step is a substance called pyruvate, which could be further broken down to yield a lot more energy, but this process is not accessible to all cells, and it turns out that the astrocytes need help for this to happen, which is where amyloid-beta comes in.
7. The Crucial Role of Amyloid-Beta
Amyloid-beta (also known as “abeta”) is the substance that forms the famous plaque that accumulates in the brains of Alzheimer’s patients. It has been believed by many (but not all) in the research community that amyloid-beta is the principal cause of Alzheimer’s, and as a consequence, researchers are actively seeking drugs that might destroy it. However, amyloid-beta has the unique capability of stimulating the production of an enzyme, lactate dehydrogenase, which promotes the breakdown of pyruvate (the product of anaerobic glucose metabolism) into lactate, through an anaerobic fermentation process, with the further production of a substantial amount of ATP.
The lactate, in turn, can be utilized itself as an energy source by some cells, and it has been established that neurons are on the short list of cell types that can metabolize lactate. So I conjecture that the lactate is transported from the astrocyte to a neighboring neuron to enhance its energy supply, thus reducing its dependence on glucose. It is also known that apoE can signal the production of amyloid-beta, but only under certain poorly understood environmental conditions. I suggest those environmental triggers have to do with the internal manufacture of fats and cholesterol as opposed to the extraction of these nutrients from the blood supply. I.e., amyloid-beta is produced as a consequence of environmental oxidative stress due to an inadequate supply of fats and cholesterol from the blood.
In addition to being utilized as an energy source by being broken down to lactate, pyruvate can also be used as a basic building block for synthesizing fatty acids. So anaerobic glucose metabolism, which yields pyruvate, is a win-win-win situation: (1) it significantly reduces the risk of exposure of fatty acids to oxygen, (2) it provides a source of fuel for neighboring neurons in the form of lactate, and (3) it provides a basic building block for fatty acid synthesis. But it depends upon amyloid-beta to work.
Thus, in my view (and in the view of others [28] [20] Amyloid-Beta and Alzheimer’s), amyloid-beta is not a cause of Alzheimer’s, but rather a protective device against it. The abstract of reference [28] arguing this point of view is reproduced in full in the Appendix. Several variants of a genetic defect associated with amyloid precursor protein (APP), the protein from which amyloid-beta is derived, have now been identified. A defect in this protein, which is associated with an increased risk of early onset Alzheimer’s, would likely lead to a reduced ability to synthesize amyloid-beta, which would then leave the brain with a big problem, since both the fuel and the basic building blocks for fatty acid synthesis would be in short supply, while oxygen trekking through the cell to the mitochondria would be exposing whatever fats were being synthesized to oxidation. The cell would likely be unable to keep up with need, and this would lead to a reduction in the number of fatty acids in the Alzheimer’s cerebrospinal fluid, a well-established characteristic of Alzheimer’s [38].
8. Cholesterol’s Role in the Brain
The brain comprises only 2% of the body’s total weight, yet it contains nearly 25% of the total cholesterol in the body. It has been determined that the limiting factor allowing the growth of synapses is the availability of cholesterol, supplied by the astrocytes. Cholesterol plays an incredibly important role in the synapse, by shaping the two cell membranes into a snug fit so that the signal can easily jump across the synapse [50]. So inadequate cholesterol in the synapse will weaken the signal at the outset, and inadequate fat coating the myelin sheath will further weaken it and slow it down during transport. A neuron that can’t send its messages is a useless neuron, and it only makes sense to prune it away and scavenge its contents.
The neurons that are damaged in Alzheimer’s are located in specific regions of the brain associated with memory and high level planning. These neurons need to transmit signals long distances between the frontal and prefrontal cortex and the hippocampus, housed in the midbrain. The transport of these signals depends on a strong and tight connection in the synapse, where the signal is transferred from one neuron to another, and a secure transmission across the long nerve fiber, a part of the white matter. The myelin sheath which coats the nerve fiber consists mainly of fatty acids, along with a substantial concentration of cholesterol. If it is not well insulated, the signal transmission rate will slow down and the signal strength will be severely reduced. Cholesterol is crucial for the myelin as well as for the synapse, as demonstrated dramatically through experiments conducted on genetically defective mice by Gesine Saher et al. [45]. These mutant mice lacked the ability to synthesize cholesterol in myelin-forming oligodendrocytes. They had severly disturbed myelin in their brains, and exhibited ataxia (uncoordinated muscle movements) and tremor. In the abstract, the authors wrote unequivocally, “This shows that cholesterol is an indispensable component of myelin membranes.”
In a post-mortem study comparing Alzheimer’s patients with a control group without Alzheimer’s, it was found that the Alzheimer’s patients had significantly reduced amounts of cholesterol, phospholipids (e.g, B-HDL), and free fatty acids in the cerebrospinal fluid than did the controls [38]. This was true irrespective of whether the Alzheimer’s patients were typed as apoE-4. In other words, reductions in these critical nutrients in the spinal fluid are associated with Alzheimer’s regardless of whether the reduction is due to defective apoE. The reductions in fatty acids were alarming: 4.5 micromol/L in the Alzheimer’s patients, compared with 28.0 micromol/L in the control group. This is a reduction by more than a factor of 6 in the amount of fatty acid available to repair the myelin sheath!
People with the apoE-4 allele tend to have high serum cholesterol. The question of whether this high cholesterol level might be an attempt on the part of the body to adjust for a poor rate of cholesterol uptake in the brain was addressed by a team of researchers in 1998 [39]. They studied 444 men between 70 and 89 years old at the time, for whom there existed extensive records of cholesterol levels dating back to several decades ago. Most significantly, cholesterol levels fell for the men who developed Alzheimer’s prior to their showing Alzheimer’s symptoms. The authors suggested that their high cholesterol might have been a protective mechanism against Alzheimer’s.
One might wonder why their cholesterol levels fell. There was no mention of statin drugs in the article, but statins would certainly be an effective way to reduce cholesterol levels. The statin industry would like people to believe that high cholesterol is a risk factor for Alzheimer’s, and they are quite thrilled that high cholesterol early in life is correlated with Alzheimer’s much later. But these results suggest quite the opposite: that blood cholesterol levels are kept high intentionally by the body regulatory mechanisms in an attempt to compensate for the defect. A high concentration will lead to an increase in the rate of delivery to the brain, where it is critically needed to keep the myelin sheath healthy and to promote neuron signaling in the synapses.
Using MRI technology, researchers at UCLA were able to measure the degree of breakdown of myelin in specific regions of the brain [6]. They conducted their studies on over 100 people between 55 and 75 years old, for whom they also determined the associated apoE allele (2, 3, or 4). They found a consistent trend in that apoE-2 had the least amount of degradation, and apoE-4 had the most, in the frontal lobe region of the brain. All of the people in the study were thus far healthy with respect to Alzheimer’s. These results show that premature breakdown of myelin sheath (likely due to an insufficient supply of fats and cholesterol to repair it) is associated with apoE-4.
To summarize, I hypothesize that, for the apoE-4 Alzheimer’s patients, defective apoE has led to an impaired ability to transport fats and cholesterol from the blood stream, via the astrocytes, into the cerebrospinal fluid. The associated high blood serum cholesterol is an attempt to partially correct for this defect. For the rest of the Alzheimer’s patients (the ones without the apoE-4 allele but who also have severely depleted fatty acids in their cerebrospinal fluid), we have to look for another reason why their fatty acid supply chain might be broken.
9. Infections and Inflammation
To summarize what I have said so far, Alzheimer’s appears to be a consequence of an inability of neurons to function properly, due to a deficiency in fats and cholesterol. A compounding problem is that the fats over time will become rancid if they cannot be adequately replenished. Rancid fats are vulnerable to attack by microorganisms such as bacteria and viruses. Amyloid-beta is part of the solution because it allows the astrocytes to be much more effective in utilizing glucose anaerobically, which protects the internally synthesized fats and cholesterol from toxic oxygen exposure, while at the same time providing the energy needed both by the astrocyte for the synthesis process and by neighboring neurons to fuel their signal firings.
Besides the astrocytes, the microglia in the brain are also implicated in Alzheimer’s. Microglia promote neuron growth when all is well, but trigger neuron programmed cell death in the presence of toxic substances secreted by bacteria such as polysaccharides [56]. Microglia will defensively secrete cytokines (communication signals that promote an immune response) when exposed to infective agents, and these in turn will lead to inflammation, another well-known feature associated with Alzheimer’s [1]. The microglia are able to control whether neurons should live or die, and they surely base this decision on factors related to how well the neuron functions and whether it is infected. Once enough neurons have been programmed for cell death, the disease will manifest itself as cognitive decline.
10. Evidence that Infection is Associated with Alzheimer’s
There is substantial evidence that Alzheimer’s is related to an increased likelihood of infective agents appearing in the brain. Some researchers believe that infective agents are the principle cause of Alzheimer’s. There are a number of bacteria that reside in the human digestive system and can co-exist with our own cells without any harm. However, H. pylori, one that is quite common, has been recently shown to be responsible for stomach ulcers. It has been suspected that H. Pylori might be implicated in Alzheimer’s, and, indeed, a recent study showed that Alzheimer’s patients had a significantly higher concentration of an antibody against H. Pylori in both their cerebrospinal fluid and their blood than non-Alzheimer’s controls [26]. H. pylori was detected in 88% of the Alzheimer’s patients but only 47% of the controls. In an effort to treat the Alzheimer’s patients, the researchers administered a potent combination of antibiotics, and assessed the degree of mental decline over the next two years [27]. For 85% of the patients, the infection was successfully routed, and for those patients, cognitive improvement was also detected after two years had elapsed. So this was a nice example of the possibility of treating Alzheimer’s through antibiotics.
C. pneumoniae is a very common bacterium, estimated to infect 40-70% of adults. But there’s a big difference between a bacterium being in the blood stream and making its way into the inner sanctum of the brain. A study of post-mortem samples from various regions of the brains of Alzheimer’s patients and non-Alzheimer’s controls revealed a remarkably different statistic: 17 out of 19 Alzheimer’s brains tested positive for the bacterium, whereas only 1 out of 19 brains from the control group tested positive [5].
Many other infective agents, both viruses and bacteria, have been found to be associated with Alzheimer’s, including herpes simplex virus, picornavirus, Borna disease virus, and spirochete [23]. One proposal was that a particularbacteriophage — a virus that infects the bacterium C. pneumoniae — might be responsible for Alzheimer’s [14]. The authors argued that the phages might make their way into the mitochondria of the host cell and subsequently initiate Alzheimer’s.
11. Ketogenic Diet as Treatment for Alzheimer’s
One of the promising new treatment paradigms for Alzheimer’s is to have the patient switch to an extremely high fat, low carb diet, a so-called “ketogenic” diet. The name comes from the fact that the metabolism of dietary fats produces “ketone bodies” as a by-product, which are a very useful resource for metabolism in the brain. It is becoming increasingly clear that defective glucose metabolism in the brain (so-called “type-3 diabetes”) is an early characteristic of Alzheimer’s. Ketone bodies, whether they enter the astrocyte directly or are produced in the astrocyte itself by breaking down fats, can be delivered to adjacent neurons, as shown in the accompanying figure.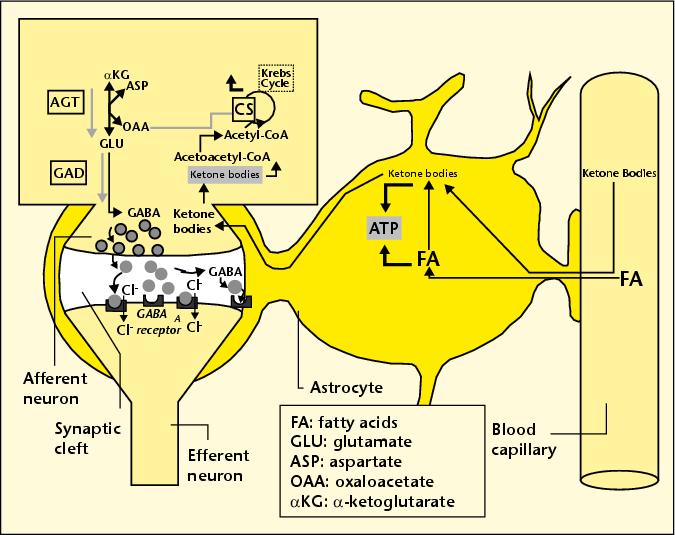 These neurons can utilize the ketone bodies both as an energy source (replacing and therefore relieving glucose) and as a precursor to GABA, a critical neurotransmitter that is widespread in the brain.
These neurons can utilize the ketone bodies both as an energy source (replacing and therefore relieving glucose) and as a precursor to GABA, a critical neurotransmitter that is widespread in the brain.
Evidence that a ketogenic diet might help Alzheimer’s was first found through research conducted on mice who had been bred to be prone to Alzheimer’s disease [21]. Researchers found that the mice’s cognition improved when they were treated with a high-fat low-carb diet, and also that the amount of amyloid-beta in their brain was reduced. The latter effect would be anticipated based on the premise that amyloid-beta promotes full utilization of glucose anaerobically, as I discussed previously. By having ketone bodies as an additional source of fuel, the dependence on glucose is reduced. But another effect that may be more important than this is the availability of high-quality fats to improve the condition of the myelin sheath.
This idea is supported by other experiments done on human Alzheimer’s patients [11] [42]. A placebo-controlled 2004 study [42] of the effect of dietary fat enrichment on Alzheimer’s is especially informative, because it uncovered a significant difference in effectiveness for the fat-enrichment for subjects who did not have the apoE-4 allele as compared with those who did. The experimental test group were given a supplemental drink containing emulsified medium chain triglycerides, found in high concentration in coconut oil. The subjects without the apoE-4 allele showed a significant improvement in score on a standard test for Alzheimer’s, whereas those with the apoE-4 allele did not. This is a strong indicator that the benefit may have to do with an increase in uptake by the astrocyte of these high-quality fats, something that the subjects with the apoE-4 allele are unable to accomplish due to the defective IDL and LDL transport mechanisms.
12. NADH Treatment: the Crucial Role of Antioxidants
One of the very few promising treatments for Alzheimer’s is the coenzyme, NADH (nicotinamide adenine dinucleotide) [12]. In a placebo-controlled study, Alzheimer’s subjects given NADH for six months exhibited significantly better performances on verbal fluency, visual constructional ability and abstract verbal reasoning than the control subjects given a placebo.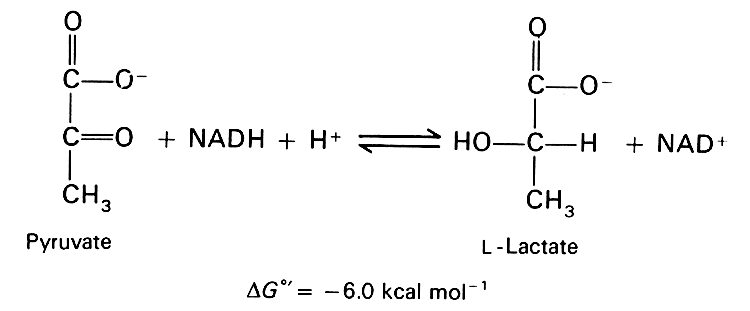
Why would NADH be effective? In the process of converting pyruvate to lactate, lactate dehydrogenase consumes oxygen by oxidizing NADH to NAD+, as illustrated in the accompanying figure. So, if the bioavailability of NADH is increased, it stands to reason that the astrocyte would have an enhanced ability to convert pyruvate to lactate, the critical step in the anaerobic metabolic pathway that is enhanced by amyloid-beta. The process, by absorbing the toxic oxygen, would reduce the damage to the lipids due to oxygen exposure, and would also provide lactate as a source of energy for the neurons.
13. Excessive Oxygen Exposure and Cognitive Decline
It has been observed that some elderly people suffer temporary and sometimes permanent cognitive decline following a lengthy operation. Researchers at the University of South Florida and Vanderbilt University suspected that this might be due to excessive exposure to oxygen [4]. Typically, during an operation, people are often administered high doses of oxygen, even as much as 100% oxygen. The researchers conducted an experiment on young adult mice, which had been engineered to be predisposed towards Alzheimer’s but had not yet suffered cognitive decline. They did however already have amyloid-beta deposits in their brains. The re-engineered mice, as well as a control group that did not have the Alzheimer’s susceptibility gene, were exposed to 100-percent oxygen for a period of three hours, three times over the course of several months, simulating repeated operations. They found that the Alzheimer’s pre-disposed mice suffered significant cognitive decline following the oxygen exposure, by contrast with the control mice.
This is a strong indication that the excessive oxygen exposure during operations is causing oxidative damage in the Alzheimer’s brain. Given the arguments I have presented above, this result makes good sense. The brain, by converting to anaerobic metabolism for generating energy (with help from amyloid-beta) is trying its best to avoid exposing the fatty acids and cholesterol to oxidative damage. But an extremely high concentration of oxygen in the blood makes it very difficult to protect the fats and cholesterol during transport through the blood, and also probably causes an unavoidable increase in oxygen uptake and therefore exposure within the brain itself.
14. Fats are a Healthy Choice!
You would practically have to be as isolated as an Australian Aborigine not to have absorbed the message that dietary fats, particularly saturated fats, are unhealthy. I am extremely confident that this message is false, but it is nearly impossible to turn the opinion tide due to its pervasive presence. Most people don’t question why fats are bad; they assume that researchers must have done their homework, and they trust the result.
To say that the current situation with regard to dietary fats is confusing would be an understatement. We are repeatedly told to keep our total fat intake down to, ideally, 20% of our total calories. This is difficult to achieve, and I believe it is misguided advice. In direct contradiction to this “low-fat” goal, we are encouraged to consume as much as possible of the “good” kinds of fats. Fortunately, the message is finally becoming widely embraced that omega-3 fats are healthy and that trans fats are extremely unhealthy. DHA (docosahexaenoic acid) is an omega-3 fat that is found in large quantities in the healthy brain. In the diet, it is available mainly from cold water fish, but eggs and dairy are also good sources. Trans fats are generated by a high-heat process that hydrolyzes polyunsaturated fats into a more stable configuration, which increases their shelf life but makes them so unnatural they almost can no longer be called a food. Trans fats are extremely damaging both to heart and brain health. A high consumption of trans fats has recently been shown to increase the risk of Alzheimer’s [41]. Trans fats are especially prevalent in highly processed foods — particularly when fats are converted to a powdered form.
We are told to avoid saturated fats, mainly because they have appeared, from empirical evidence, to be more likely to raise LDL levels than unsaturated fats. Yet these fats are less susceptible to oxidation, and this may be why they show up in LDL — because they are of higher quality and therefore should preferentially be delivered to the tissues for functional roles rather than as fuel (i.e., free fatty acids). Coconut oil, a saturated fat, has been shown to benefit Alzheimer’s patients [42]. And high-fat dairy (also highly saturated) has been shown to be beneficial both to fertility among women [10] and, remarkably, to heart disease [37][22].
Despite the widespread belief that fats (particularly saturated fats) are unhealthy, an article that appeared in the American Journal of Clinical Nutrition in 2004 [37] claims that, for a group of post-menopausal women, a high-fat, high-saturated-fat diet affords better protection from coronary artery disease than a low-fat (25% of calories from fats) diet. The subjects in the study were obese women with coronary artery disease. Most of them had high blood pressure, and many had diabetes. They fit the profile for metabolic syndrome that I have previously argued is a direct consequence of a prolonged low-fat high-carb diet. I am gratified to see that my hypothesis that an increase in fat intake would decrease their risk of heart disease has been verified by a carefully controlled study.
Another investigation where fats were shown to afford protection against heart disease has just been completed. It involved a long-term study of a large number of Swedish men [22]. The authors looked at low- vs high-fat dairy, as well as consumption of fruits and vegetables, meats, grains, etc. The only statistically significant result that afforded protection from heart disease was a combination of high-fat dairy and lots of fruits and vegetables. Fruits and vegetables with low-fat dairy afforded no protection.
I suspect one of the critical nutrients the fruits and vegetables provide is antioxidants that help prolong the life of the fats. Other excellent sources of antioxidants include richly colored fruits like berries and tomatoes, coffee, green tea, and dark chocolate, and several spices, most especially cinnamon and turmeric (a major ingredient of curry). These should be consumed in abundance along with fats for optimal results.
Polyunsaturated fats such as corn oil and canola oil are unhealthy for the brain precisely because they are unsaturated. There are two major problems: (1) they have a low melting point, which means that, if they are used for frying they will be converted to trans fats, which are extremely unhealthy, and (2) they are much more susceptible to becoming rancid (oxidized) at room temperature than saturated fats, i.e., they have a shorter shelf life.
Researchers in Germany recently conducted an ingenious experiment designed to determine how the degree of freshness of polyunsaturated fats affects the metabolism of those fats in female lactating rats [43]. They divided female rats into two groups, and the only difference between the test group and the controls was that the test group was given fats that had been left in a relatively warm place for 25 days, which caused considerable oxidative damage, whereas the controls were fed fresh fats instead. The rats’ unusual diet was begun on the day that they gave birth to a litter. The researchers examined the mammary glands and the milk produced by the two groups for apparent differences. They found that the test group’s milk was markedly reduced in the amount of fat it contained, and their mammary glands correspondingly took up less fat from the blood supply. One might surmise that the rats’ metabolic mechanisms were able to detect oxidative damage to the fats, and therefore rejected them, prefering to do without rather than to risk the consequences of feeding their pups oxidized fats. Consequently, the pups of the test group gained significantly less weight than the control group’s pups.
Boxed items like cookies and crackers that contain processed polyunsaturated fats are doctored with antioxidants and even antibiotics to protect them from spoiling. Once they’re consumed, however, they still have to be protected from going rancid. Biochemical laws work the same way whether inside or outside the body. There are plenty of bacteria throughout the body that would be eager to take up house-keeping in rancid fats. The body has devised all kinds of strategies for protecting fats from oxidation (becoming rancid) and from attack by bacteria. But its task is rendered much easier for saturated rather than unsaturated fats, and for fresh rather than stale fats.
If we stop trying to get by on as few fats as possible in the diet, then we don’t have to become so preoccupied with getting the “right” kinds of fats. If the body is supplied with an overabundance of fats, it can pick and choose to find the perfect fat to match each particular need; excess or defective fats can just be used as fuel, where it’s not very important which fat it is, as long as it can be broken down to release energy.
15. Summary and Conclusion
This is an exciting time for Alzheimer’s research, as new and surprising discoveries are coming out at a rapid pace, and evidence is mounting to support the notion that Alzheimer’s is a nutritional deficiency disease. It is an indication of how much progress has been made in recent years to note that 42% of the references in this essay were published in 2008 or 2009. A popular new theory is that Alzheimer’s may grow out of an impaired ability to metabolize glucose in the brain. The term “type-3 diabetes” has been coined to describe this defect, which often appears long before any symptoms of Alzheimer’s [49]. A shift from aerobic towards anaerobic glucose metabolism in the brain seems to be a harbinger of Alzheimer’s later in life, but I argue that the reason for this shift is both to provide a basic ingredient (pyruvate) from which to synthesize fatty acids, while simultaneously protecting them from potentially damaging oxidation. The ApoE-4 allele, which is associated with increased risk to Alzheimer’s, clearly implicates defects in fat and cholesterol transport, and the remarkable 6-fold reduction in the amount of fatty acids present in the cerebrospinal fluid of Alzheimer’s patients [38] speaks loudly the message that fat insufficiency is a key part of the picture. The observation that the myelin is degraded in the frontal lobes of the brains of people possessing the apoE-4 allele further substantiates the theory that the myelin repair mechanism is defective.
Cholesterol obviously plays a vital role in brain function. A whopping 25% of the total cholesterol in the body is found in the brain, and it is present in abundance both in the synapses and in the myelin sheath. The cholesterol in both of these places has been shown to play an absolutely essential role in signal transport and in growth and repair.
Given the strong positive role played by cholesterol, it can only be assumed that statin drugs would increase the risk of developing Alzheimer’s. However, the statin industry has been remarkably successful thus far in hiding this painful fact. They have managed to make much of the observation that high cholesterol much earlier in life is associated with an increased risk to Alzheimer’s thirty years later. Yet they offer not a single study, not even a retrospective study, to substantiate any claim that actively reducing cholesterol through statin therapy would improve the situation for these people. In fact, most damningly, the statin usage evidence that would answer the question was “unavailable” to the researchers who conducted the seminal study.
Beatrice Golomb is an M.D. Ph.D. who heads up the UCSD Statin Study group, a research team who are actively investigating the risk-benefit balance of statin drugs. She is increasingly becoming convinced that statin drugs should not be recommended for the elderly: that in their case the risks clearly outweigh the benefits. She makes a strong case for this position in an on-line article available here [15]. The section on Alzheimer’s is particularly compelling, and it points out the pitfalls in relying on previous studies done by the statin industry, where often those who have memory problems as side-effects of the statin drugs are excluded from the study, so that the results end up inappropriately biased in favor of statins. In summary, she wrote: “It must be emphasized that the randomized trial evidence has, to date, uniformly failed to show cognitive benefits by statins and has supported no effect or frank and significant harm to cognitive function.”
In addition to refusing to take statin therapy, another way in which an individual can improve their odds against Alzheimer’s is to consume plenty of dietary fats. It seems odd to suddenly switch from a “healthy” low-fat diet to an extremely high fat ketogenic diet, once a diagnosis of Alzheimer’s is made. A ketogenic diet consists, ideally, of 88% fat, 10% protein, and 2% carbohydrate [11]. That is to say, it is absurdly high in fat content. It seems much more reasonable to aim for something like 50% fat, 30% protein, and 20% carbohydrate, so as to pro-actively defend against Alzheimer’s.
I highly recommend a recent book written by the pediatric brain surgeon, Larry McCleary, M.D., called The Brain Trust Program [33]. This book gives a wealth of fascinating information about the brain, as well as specific recommendations for ways to improve cognitive function and avert later Alzheimer’s. Most significantly, he recommends a diet that is high in cholesterol and animal fats, including an abundance of fish, seafood, meat, and eggs. He also recommends coconuts, almonds, avocados and cheese, all foods that contain a significant amount of fat, while encouraging the avoidance of “empty carbs.” His knowledge on this subject grew out of his interest in helping his young patients heal more rapidly after brain trauma.
Our nation is currently bracing itself for an onslaught of Alzheimer’s, at a time when baby boomers are approaching retirement, and our health care system is already in a crisis of escalating costs and shrinking funds. We can not afford the high cost of caring for the swelling population of Alzheimer’s patients that our current practices of low-fat diet and ever expanding statin usage are promoting.
Appendix In this appendix, I include the full abstract of two papers that are relevant to the theory presented here. The first is the abstract of reference [19] in [46], which is reference [44] here [see the section on statin drugs above for context]:
Abstract, “Epidemiological and clinical trials evidence about a preventive role for statins in Alzheimer’s disease:”
“This paper reviews epidemiological and clinical trials data about whether statin use reduces the risk of Alzheimer’s disease (AD). The available information has come in three waves. The initial, mostly cross-sectional observational reports suggested that statins might prevent dementia. Next, two large clinical trials with cognitive add-on studies showed no benefit and neither did the third wave, again with observational studies. The latter were mostly longitudinal, and were critical of the first studies for not adequately addressing confounding by indication (i.e. that patients with dementia would be denied statins). Most recently, new data from the Canadian Study of Health and Aging have produced a mixed result. While methodological considerations are clearly important in understanding why the reports are so variable, there might also be merit in differentiating between statins, based on their presumed – and variable – mechanisms of action in dementia prevention, before concluding that the initial reports are entirely artefactual. Still, the first reports appear to have overestimated the extent of protection, so that unless there are important effects achievable with specific statins, a more than a modest role for statins in preventing AD seems unlikely.” The second abstract is taken from reference [28], on the “alternative hypothesis” that amyloid-beta is protective rather than detrimental to Alzheimer’s, i.e., that it is a “protective response to neuronal insult:”
Abstract, “Amyloid-beta in Alzheimer disease: the null versus the alternate hypotheses:”
“For nearly 20 years, the primary focus for researchers studying Alzheimer disease has been centered on amyloid-beta, such that the amyloid cascade hypothesis has become the “null hypothesis.” Indeed, amyloid-beta is, by the current definition of the disease, an obligate player in pathophysiology, is toxic to neurons in vitro, and, perhaps most compelling, is increased by all of the human genetic influences on the disease. Therefore, targeting amyloid-beta is the focus of considerable basic and therapeutic interest. However, an increasingly vocal group of investigators are arriving at an “alternate hypothesis” stating that amyloid-beta, while certainly involved in the disease, is not an initiating event but rather is secondary to other pathogenic events. Furthermore and perhaps most contrary to current thinking, the alternate hypothesis proposes that the role of amyloid-beta is not as a harbinger of death but rather a protective response to neuronal insult. To determine which hypothesis relates best to Alzheimer disease requires a broader view of disease pathogenesis and is discussed herein.”
Contact: seneff@csail.mit.edu
References
[1] H. Akiyama, S. Barger, S. Barnum, B. Bradt, J.Bauer, G.M. Cole, N.R. Cooper, P. Eikelenboom, M. Emmerling, B.L. Fiebich, C.E. Finch, S. Frautschy, W.S. Griffin, H. Hampel, M. Hull, G. Landreth, L. Lue, R. Mrak, I.R. Mackenzie, P.L. McGeer, M.K. O’Banion, J. Pachter, G. Pasinetti, C. Plata-Salaman, J. Rogers, R.Rydel, Y. Shen, W. Streit, R. Strohmeyer, I. Tooyoma, F.L. Van Muiswinkel, R. Veerhuis, D. Walker, S. Webster, B. Wegrzyniak, G. Wenk, and T. Wyss-Coray, “Inflammation and Alzheimer’s disease.” Neurobiol Aging (2000) May-Jun;21(3):383-421,
[2] Alzheimer’s Association, “Alzheimer’s Disease Facts and Figures,” Alzheimer’s and Dementia (2009) Vol. 5, Issue 3.
[3] K.J. Anstey, D.M. Lipnicki and L.F. Low, “Cholesterol as a risk factor for dementia and cognitive decline: a systematic review of prospective studies with meta-analysis.” Am J Geriatr Psychiatry (2008) May, Vol. 16, No. 5, pp. 343-54.
[4] G. Arendash, A. Cox, T. Mori, J. Cracchiolo, K. Hensley, J. Roberts 2nd, “Oxygen treatment triggers cognitive impairment in Alzheimer’s transgenic mice,” Neuroreport. (2009) Jun 18.
[5] B.J. Balin, C.S. Little, C.J. Hammond, D.M. Appelt, J.A. Whittum-Hudson, H.C. Gerard, A.P. Hudson, “Chlamydophila pneumoniae and the etiology of late-onset Alzheimer’s disease.” J. Alz. Dis. (2008) Vol. 13, pp. 371-380.
[6] G. Bartzokis, MD; P.H. Lu, Psy, D.H. Geschwind, MD, N.Edwards, MA, J. Mintz, PhD, and J.L. Cummings, MD, “Apolipoprotein E Genotype and Age-Related Myelin Breakdown in Healthy Individuals: Implications for Cognitive Decline and Dementia,” Arch Gen Psychiatry (2006) Vol. 63, pp. 63-72.
[7] N. Bernoud, L. Fenart, C. Bénistant, J. F. Pageaux, M. P. Dehouck, P. Molière, M. Lagarde, R. Cecchelli,d, and J. Lecerf, “Astrocytes are mainly responsible for the polyunsaturated fatty acid enrichment in blood-brain barrier endothelial cells in vitro” Journal of Lipid Research (1998) Sept., Vol. 39, pp. 1816-1824.
[8] M. S. Brown and J. L. Goldstein, “A Receptor-Mediated Pathway for Cholesterol Homeostasis,” Nobel Lecture, December 9, 1985.
[9] N. Cartier, C. Sevin, A. Benraiss, P. DeDeyn, D. Bonnin, M-T Vanier, M. Philippe, V. Gieselmann and P. Aubourg, “AAV5-Mediated Delivery of Human Aryl Sulfatase A (hARSA) Prevents Sufatide Storage and Neuropathological Phenotype in Metachromatic Leukodystrophy (MLD) Mice,” Molecular Therapy (2005) 11, S166-S167; doi: 10.1016/j.ymthe.2005.06.431
[10] J. Chavarro, W.C. Willett, and P.J. Skerrett, The Fertility Diet, (2008) McGraw Hill.
[11] L.C. Costantini, L.J. Barr, J.L. Vogel and S.T. Henderson, “Hypometabolism as a therapeutic target in Alzheimer’s disease” BMC Neurosci (2008) Vol. 9, Suppl. 2, S16. doi: 10.1186/1471-2202-9-S2-S16.
[12] V. Demarin, S.S. Podobnik, D. Storga-Tomic and G. Kay, “Treatment of Alzheimer’s disease with stabilized oral nicotinamide adenine dinucleotide: A randomized, double-blind study” Drugs Exp Clin Res. (2004) Vol. 30, No. 1, pp. 27-33.
[13] R.B. DeMattos, R.P. Brendza, J.E. Heuser, M.Kierson, J.R. Cirrito, J. Fryer, P.M. Sullivan, A.M. Fagan, X. Han and D.M. Holtzman, “Purification and characterization of astrocyte-secreted apolipoprotein E and J-containing lipoproteins from wild-type and human apoE transgenic mice,” Neurochem Int. (2001) Nov-Dec;39(5-6):415-25. doi:10.1016/S0197-0186(01)00049-3.
[14] M. Dezfulian, M.A. ShokrgozarA, S. Sardari, K. Parivar and G. Javadi, “Can phages cause Alzheimer’s disease?” Med Hypotheses (2008) Nov;71(5):651-6.
[15] B.A. Golomb, M.D., Ph.D., “Statin Adverse Effects: Implications for the Elderly,” Geriatric Times (2004) May/June, Vol. V, Issue 3
[16] W.R. Grant, Ph.D., “Does Vitamin D Reduce the Risk of Dementia?” Journal of Alzheimer’s Disease (2009) May, Vol. 17, No. 1., pp. 151-9.
[17] Dr. Duane Graveline, Lipitor: Thief of Memory, Statin Drugs and the Misguided War on Cholesterol, (2004) http://www.buybooksontheweb.com.
[18] X. Han, “Potential mechanisms contributing to sulfatide depletion at the earliest clinically recognizable stage of Alzheimer’s disease: a tale of shotgun lipidomics,” J Neurochem (2007) November, Vol. 103, Suppl. 1. pp. 171-179. doi: 10.1111/j.1471-4159.2007.04708.x.
[19] X. Han, H. Cheng, J.D. Fryer, A.M. Fagan and D.M. Holtzman, “Novel Role for Apolipoprotein E in the Central Nervous System: Modulation of Sulfatide Content” Journal of Biological Chemistry, March 7, 2003, Vol. 278, pp. 8043-8051, DOI 10.1074/jbc.M212340200.
[20] K. Heininger, “A unifying hypothesis of Alzheimer’s disease. IV. Causation and sequence of events,” Rev Neurosci. (2000) Vol. 11, Spec No, pp.213-328.
[21] S.T. Henderson, “Ketone Bodies as a Therapeutic for Alzheimer’s Disease,” NeuroTherapeutics,, (2008) Jul;5(3):470-80, doi:10.1016/j.nurt.2008.05.004
[22] S. Holmberg, A. Thelin and E.-L. StiernstrNvm, “Food Choices and Coronary Heart Disease: A Population Based Cohort Study of Rural Swedish Men with 12 Years of Follow-up,” Int. J. Environ. Res. Public Health (2009) Vol. 6, pp. 2626-2638;
[23] K. Honjo, R. van Reekum, and N.P. Verhoeff, “Alzheimer’s disease and infection: do infectious agents contribute to progression of Alzheimer’s disease?” Alzheimers Dement. (2009) Jul;5(4):348-60.
[24] S.M. Innis and R.A. Dyer, “Brain astrocyte synthesis of docosahexaenoic acid from n-3 fatty acids is limited at the elongation of docosapentaenoic acid,” (2002) Sept. Journal of Lipid Research, Vol. 43, pp. 1529-1536.
[25] L. Jeng, A.V. Yamshchikov, S.E. Judd, H.M. Blumberg, G.S. Martin, T.R. Ziegler and V. Tangpricha, “Alterations in Vitamin D Status and Anti-microbial Peptide Levels in Patients in the Intensive Care Unit with Sepsis,” Journal of translational Medicine,” (2009) Vol. 7, No. 28.
[26] J. Kountouras, M. Boziki, E. Gavalas, C. Zavos, G. Deretzi, N. Grigoriadis, M. Tsolaki, D. Chatzopoulos, P. Katsinelos, D. Tzilves, A. Zabouri, I. Michailidou, “Increased cerebrospinal fluid Helicobacter pylori antibody in Alzheimer’s disease,” Int J Neurosci. (2009) 119(6):765-77.
[27] J. Kountouras, M. Boziki, E. Gavalas, C. Zavos, N. Grigoriadis, G. Deretzi, D. Tzilves, P. Katsinelos, M. Tsolaki, D. Chatzopoulos, and I. Venizelos, “Eradication of Helicobacter pylori may be beneficial in the management of Alzheimer’s disease,” J Neurol. (2009) May;256(5):758-67. Epub 2009 Feb 25.
[28] H.G. Lee, X. Zhu, R.J. Castellani, A. Nunomura, G. Perry, and M.A. Smith, “Amyloid-beta in Alzheimer disease: the null versus the alternate hypotheses,” J Pharmacol Exp Ther. (2007) June, Vol. 321 No. 3, pp. 823-9. doi:10.3390/ijerph6102626.
[29] J. Marcus, S. Honigbaum, S. Shroff, K. Honke, J. Rosenbluth and J.L. Dupree, “Sulfatide is essential for the maintenance of CNS myelin and axon structure,” Glia (2006), Vol. 53, pp. 372-381.
[30] R.T. Matthews, L. Yang, S. Browne, M. Baik and M.F. Beal, “Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects,” Proc Natl Acad Sci U S A. (1998) Jul 21, Vol. 95, No. 15, pp.8892-7.
[31] D. Lutjohann and K. von Bergmann, “24S-hydroxycholesterol: a marker of brain cholesterol metabolism”Pharmacopsychiatry (2003) January 10, Vol. 36 Suppl 2, pp. S102-6, DOI: 10.1055/s-2003-43053.
[32] J. C. McCann and B.N. Ames, “Is there convincing biological or behavioral evidence linking vitamin D deficiency to brain dysfunction?”, (2008) FASEB J. Vol. 22, pp. 982-1001. doi: 10.1096/fj.07-9326rev.
[33] Larry McCleary, M.D., The Brain Trust Program (2007) September, The Penguin Group, New York, New York.
[34] B. McGuinness et al., “Statins for the prevention of dementia,” Cochrane Database of Systematic Reviews,(2009) No. 2.
[35] M.M. Mielke, P.P. Zandi, M. Sjogren, et al. “High total cholesterol levels in late life associated with a reduced risk of dementia,” Neurology (2005) Vol. 64, pp. 1689-1695.
[36] S.A. Moore, “Polyunsaturated Fatty Acid Synthesis and Release by Brain-Derived Cells in Vitro,” Journal of Molecular Neuroscience (2001), Vol. 16, pp. 195ff.
[37] D. Mozaffarian, E.B. Rimm, D.M. Herrington, “Dietary fats, carbohydrate, and progression of coronary atherosclerosis in postmenopausal women,” Am J Clin Nutr (2004) Vol. 80, pp. 1175-84.
[38] M. Mulder, R. Ravid, D.F. Swaab, E.R. de Kloet, E.D. Haasdijk, J. Julk, J.J. van der Boom and L.M. Havekes, “Reduced levels of cholesterol, phospholipids, and fatty acids in cerebrospinal fluid of Alzheimer disease patients are not related to apolipoprotein E4,” Alzheimer Dis Assoc Disord. (1998) Sep, Vol. 12, No. 3, pp. 198-203.
[39] I.L. Notkola, R. Sulkava, J. Pekkanen, T. Erkinjuntti, C. Ehnholm, P. Kivinen, J. Tuomilehto, and A. Nissinen, “Serum total cholesterol, apolipoprotein E epsilon 4 allele, and Alzheimer’s disease,” Neuroepidemiology (1998) Vol. 17, No. 1, pp. 14-20.
[40] F.W. Pfrieger, “Outsourcing in the brain: Do neurons depend on cholesterol delivery by astrocytes?”, BioEssays(2003) Vol. 25 Issue 1, pp.72-78.
[41] A. Phivilay, C. Julien, C. Tremblay, L. Berthiaume, P. Julien, Y. Giguère and F. Calon, “High dietary consumption of trans fatty acids decreases brain docosahexaenoic acid but does not alter amyloid-beta and tau pathologies in the 3xTg-AD model of Alzheimer’s disease.” Neuroscience (2009) Mar 3, Vol. 159, No. 1, pp. 296-307. Epub 2008 Dec 14.
[42] M.A. Reger, S. T. Henderson, C. Hale, B. Cholerton, L.D. Baker, G.S. Watson, K. Hyde, D. Chapman and S. Craft, “Effects of Beta-hydroxybutyrate on cognition in memory-impaired adults,” Neurobiology of Aging (2004) Vol. 25, No. 3, March, pp. 311-314,
[43] R. Ringseis, C. Dathe, A. Muschick, C. Brandsch and K. Eder, “Nutrient Physiology, Metabolism, and Nutrient-Nutrient Interactions Oxidized Fat Reduces Milk Triacylglycerol Concentrations by Inhibiting Gene Expression of Lipoprotein Lipase and Fatty Acid Transporters in the Mammary Gland of Rats,” American Society for Nutrition J. Nutr. (2007) Sept., Vol. 137, pp. 2056-2061.
[44] K. Rockwood, “Epidemiological and clinical trials evidence about a preventive role for statins in Alzheimer’s disease.” Acta Neurol Scand Suppl. (2006) Vol. 185, pp. 71-7.
[45] G. Saher, B. Brugger, C. Lappe-Siefke, W. Mobius, R. Tozawa, M.C. Wehr, F. Wieland, S. Ishibashi, and K.A. Nave, “High cholesterol level is essential for myelin membrane growth.” Nat Neurosci (2005) Apr, Vol. 8, No. 4, pp. 468-75. Epub 2005 Mar 27.
[46] A. Solomon, M. Kivipelto, B. Wolozin, J. Zhou, and R.A. Whitmer, “Midlife Serum Cholesterol and Increased Risk of Alzheimer’s and Vascular Dementia Three Decades Later,” Dementia and Geriatric Cognitive Disorders (2009) Vol. 28, pp. 75-80, DOI: 10:1159/000231980.
[47] M. Simons, MD, P. Keller, PhD, J. Dichgans, MD and J.B. Schulz, MD, “Cholesterol and Alzheimer’s disease: Is there a link?” Neurology (2001) Vol. 57, pp. 1089-1093.
[48] L.L. Smith, “Another cholesterol hypothesis: cholesterol as antioxidant,” Free Radic Biol Med. (1991) Vol. 11, No. 1, pp. 47-61.
[49] E. Steen, B.M. Terry, E.J. Rivera, J.L. Cannon, T.R. Neely, R. Tavares, X.J. Xu, J.R. Wands, and S.M. de la Monte “Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease – is this type 3 diabetes?” Journal of Alzheiner’s Disease (2005) Vol. 7, Number 1, pp. 63-80.
[50] J. Tong, P.P. Borbat, J.H. Freed and Y-K Shin, “A scissors mechanism for stimulation of SNARE-mediated lipid mixing by cholesterol,” PNAS (2009) March 31 Vol. 106, No. 13, pp. 5141-5146.
[51] M-C Vohl, T. A.-M. Neville, R. Kumarathasan, S. Braschi, and D.L. Sparks, “A Novel Lecithin-Cholesterol Acyltransferase Antioxidant Activity Prevents the Formation of Oxidized Lipids during Lipoprotein Oxidation,”Biochemistry (1999) Vol. 38 No. 19, pp. 5976-5981. DOI: 10.1021/bi982258w.
[52] M. Waldman, MD,, 9th International Conference on Alzheimer’s and Parkinson’s Diseases (2009) Abstract 90, Presented March 12-13.
[53] R. West, M.A., M. Schnaider Beeri, Ph.D., J. Schmeidler, Ph.D., C. M. Hannigan, B.S., G. Angelo, M.S., H.T. Grossman, M.D., C. Rosendorff, M.D., Ph.D., and J.M. Silverman, Ph.D., “Better memory functioning associated with higher total and LDL cholesterol levels in very elderly subjects without the APOE4 allele,” Am J Geriatr Psychiatry(2008) September; Vol. 16, No. 9, pp. 781-785. doi: 10.1097/JGP.0b013e3181812790.
[54] A.W.E. Weverling-Rijnsburger, G.J. Blauw, A.M. Lagaay, D.L. Knook, A.E. Meinders, and R.G.J. Westendorp, “Total cholesterol and risk of mortality in the oldest old,” The Lancet, (1997) Vol. 350, No. 9085, pp. 1119-1123,
[55] R.F. Wilson, J.F. Barletta and J.G. Tyburski, “Hypocholesterolemia in Sepsis and Critically Ill or Injured Patients”Critical Care (2003), Vol. 7, pp. 413-414.
[56] S.-C. Zhang and S. Fedoroff, “Neuron-microglia Interactions in Vitro,” Acta Neuropathol (1996) Vol. 91, pp. 385-395.
How Statins Really Work Explains Why They Don’t Really Work
By Stephanie Seneff | March 11, 2011
Introduction
The statin industry has enjoyed a thirty year run of steadily increasing profits, as they find ever more ways to justify expanding the definition of the segment of the population that qualify for statin therapy. Large, placebo-controlled studies have provided evidence that statins can substantially reduce the incidence of heart attack. High serum cholesterol is indeed correlated with heart disease, and statins, by interfering with the body’s ability to synthesize cholesterol, are extremely effective in lowering the numbers. Heart disease is the number one cause of death in the U.S. and, increasingly, worldwide. What’s not to like about statin drugs?
I predict that the statin drug run is about to end, and it will be a hard landing. The thalidomide disaster of the 1950’s and the hormone replacement therapy fiasco of the 1990’s will pale by comparison to the dramatic rise and fall of the statin industry. I can see the tide slowly turning, and I believe it will eventually crescendo into a tidal wave, but misinformation is remarkably persistent, so it may take years.
I have spent much of my time in the last few years combing the research literature on metabolism, diabetes, heart disease, Alzheimer’s, and statin drugs. Thus far, in addition to posting essays on the web, I have, together with collaborators, published two journal articles related to metabolism, diabetes, and heart disease (Seneff1 et al., 2011), and Alzheimer’s disease (Seneff2 et al., 2011). Two more articles, concerning a crucial role for cholesterol sulfate in metabolism, are currently under review (Seneff3 et al., Seneff4 et al.). I have been driven by the need to understand how a drug that interferes with the synthesis of cholesterol, a nutrient that is essential to human life, could possibly have a positive impact on health. I have finally been rewarded with an explanation for an apparent positive benefit of statins that I can believe, but one that soundly refutes the idea that statins are protective. I will, in fact, make the bold claim that nobody qualifies for statin therapy, and that statin drugs can best be described as toxins.
Cholesterol and Statins
I would like to start by reexamining the claim that statins cut heart attack incidence by a third. What exactly does this mean? A meta study reviewing seven drug trials, involving in total 42,848 patients, ranging over a three to five year period, showed a 29% decreased risk of a major cardiac event (Thavendiranathan et al., 2006). But because heart attacks were rare among this group, what this translates to in absolute terms is that 60 patients would need to be treated for an average of 4.3 years to protect one of them from a single heart attack. However, essentially all of them will experience increased frailty and mental decline, a subject to which I will return in depth later on in this essay.
The impact of the damage due to the statin anti-cholesterol mythology extends far beyond those who actually consume the statin pills. Cholesterol has been demonized by the statin industry, and as a consequence Americans have become conditioned to avoid all foods containing cholesterol. This is a grave mistake, as it places a much bigger burden on the body to synthesize sufficient cholesterol to support the body’s needs, and it deprives us of several essential nutrients. I am pained to watch someone crack open an egg and toss out the yolk because it contains “too much” cholesterol. Eggs are a very healthy food, but the yolk contains all the important nutrients. After all, the yolk is what allows the chick embryo to mature into a chicken. Americans are currently experiencing widespread deficiencies in several crucial nutrients that are abundant in foods that contain cholesterol, such as choline, zinc, niacin, vitamin A and vitamin D.
Cholesterol is a remarkable substance, without which all of us would die. There are three distinguishing factors which give animals an advantage over plants: a nervous system, mobility, and cholesterol. Cholesterol, absent from plants, is the key molecule that allows animals to have mobility and a nervous system. Cholesterol has unique chemical properties that are exploited in the lipid bilayers that surround all animal cells: as cholesterol concentrations are increased, membrane fluidity is decreased, up to a certain critical concentration, after which cholesterol starts to increase fluidity (Haines, 2001). Animal cells exploit this property to great advantage in orchestrating ion transport, which is essential for both mobility and nerve signal transport. Animal cell membranes are populated with a large number of specialized island regions appropriately called lipid rafts. Cholesterol gathers in high concentrations in lipid rafts, allowing ions to flow freely through these confined regions. Cholesterol serves a crucial role in the non-lipid raft regions as well, by preventing small charged ions, predominantly sodium (Na+) and potassium (K+), from leaking across cell membranes. In the absence of cholesterol, cells would have to expend a great deal more energy pulling these leaked ions back across the membrane against a concentration gradient.
In addition to this essential role in ion transport, cholesterol is the precursor to vitamin D3, the sex hormones, estrogen, progesterone, and testosterone, and the steroid hormones such as cortisol. Cholesterol is absolutely essential to the cell membranes of all of our cells, where it protects the cell not only from ion leaks but also from oxidation damage to membrane fats. While the brain contains only 2% of the body’s weight, it houses 25% of the body’s cholesterol. Cholesterol is vital to the brain for nerve signal transport at synapses and through the long axons that communicate from one side of the brain to the other. Cholesterol sulfate plays an important role in the metabolism of fats via bile acids, as well as in immune defenses against invasion by pathogenic organisms.
Statin drugs inhibit the action of an enzyme, HMG coenzyme A reductase, that catalyses an early step in the 25-step process that produces cholesterol. This step is also an early step in the synthesis of a number of other powerful biological substances that are involved in cellular regulation processes and antioxidant effects. One of these is coenzyme Q10, present in the greatest concentration in the heart, which plays an important role in mitochondrial energy production and acts as a potent antioxidant (Gottlieb et al., 2000). Statins also interfere with cell-signaling mechanisms mediated by so-called G-proteins, which orchestrate complex metabolic responses to stressed conditions. Another crucial substance whose synthesis is blocked is dolichol, which plays a crucial role in the endoplasmic reticulum. We can’t begin to imagine what diverse effects all of this disruption, due to interference with HMG coenzyme A reductase, might have on the cell’s ability to function.
LDL, HDL, and Fructose
We have been trained by our physicians to worry about elevated serum levels of low density lipoprotein (LDL), with respect to heart disease. LDL is not a type of cholesterol, but rather can be viewed as a container that transports fats, cholesterol, vitamin D, and fat-soluble anti-oxidants to all the tissues of the body. Because they are not water-soluble, these nutrients must be packaged up and transported inside LDL particles in the blood stream. If you interfere with the production of LDL, you will reduce the bioavailability of all these nutrients to your body’s cells.
The outer shell of an LDL particle is made up mainly of lipoproteins and cholesterol. The lipoproteins contain proteins on the outside of the shell and lipids (fats) in the interior layer. If the outer shell is deficient in cholesterol, the fats in the lipoproteins become more vulnerable to attack by oxygen, ever-present in the blood stream. LDL particles also contain a special protein called “apoB” which enables LDL to deliver its goods to cells in need. ApoB is vulnerable to attack by glucose and other blood sugars, especially fructose. Diabetes results in an increased concentration of sugar in the blood, which further compromises the LDL particles, by gumming up apoB. Oxidized and glycated LDL particles become less efficient in delivering their contents to the cells. Thus, they stick around longer in the bloodstream, and the measured serum LDL level goes up.
Worse than that, once LDL particles have finally delivered their contents, they become “small dense LDL particles,” remnants that would ordinarily be returned to the liver to be broken down and recycled. But the attached sugars interfere with this process as well, so the task of breaking them down is assumed instead by macrophages in the artery wall and elsewhere in the body, through a unique scavenger operation. The macrophages are especially skilled to extract cholesterol from damaged LDL particles and insert it into HDL particles. Small dense LDL particles become trapped in the artery wall so that the macrophages can salvage and recycle their contents, and this is the basic source of atherosclerosis. HDL particles are the so-called “good cholesterol,” and the amount of cholesterol in HDL particles is the lipid metric with the strongest correlation with heart disease, where less cholesterol is associated with increased risk. So the macrophages in the plaque are actually performing a very useful role in increasing the amount of HDL cholesterol and reducing the amount of small dense LDL.
The LDL particles are produced by the liver, which synthesizes cholesterol to insert into their shells, as well as into their contents. The liver is also responsible for breaking down fructose and converting it into fat (Collison et al., 2009). Fructose is ten times more active than glucose at glycating proteins, and is therefore very dangerous in the blood serum (Seneff1 et al., 2011). When you eat a lot of fructose (such as the high fructose corn syrup present in lots of processed foods and carbonated beverages), the liver is burdened with getting the fructose out of the blood and converting it to fat, and it therefore can not keep up with cholesterol supply. As I said before, the fats can not be safely transported if there is not enough cholesterol. The liver has to ship out all that fat produced from the fructose, so it produces low quality LDL particles, containing insufficient protective cholesterol. So you end up with a really bad situation where the LDL particles are especially vulnerable to attack, and attacking sugars are readily available to do their damage.
How Statins Destroy Muscles
Europe, especially the U.K., has become much enamored of statins in recent years. The U.K. now has the dubious distinction of being the only country where statins can be purchased over-the-counter, and the amount of statin consumption there has increased more than 120% in recent years (Walley et al, 2005). Increasingly, orthopedic clinics are seeing patients whose problems turn out to be solvable by simply terminating statin therapy, as evidenced by a recent report of three cases within a single year in one clinic, all of whom had normal creatine kinase levels, the usual indicator of muscle damage monitored with statin usage, and all of whom were “cured” by simply stopping statin therapy (Shyam Kumar et al., 2008). In fact, creatine kinase monitoring is not sufficient to assure that statins are not damaging your muscles (Phillips et al., 2002).
Since the liver synthesizes much of the cholesterol supply to the cells, statin therapy greatly impacts the liver, resulting in a sharp reduction in the amount of cholesterol it can synthesize. A direct consequence is that the liver is severely impaired in its ability to convert fructose to fat, because it has no way to safely package up the fat for transport without cholesterol (Vila et al., 2011). Fructose builds up in the blood stream, causing lots of damage to serum proteins.
The skeletal muscle cells are severely affected by statin therapy. Four complications they now face are: (1) their mitochondria are inefficient due to insufficient coenzyme Q10, (2) their cell walls are more vulnerable to oxidation and glycation damage due to increased fructose concentrations in the blood, reduced choleserol in their membranes, and reduced antioxidant supply, (3) there’s a reduced supply of fats as fuel because of the reduction in LDL particles, and (4) crucial ions like sodium and potassium are leaking across their membranes, reducing their charge gradient. Furthermore, glucose entry, mediated by insulin, is constrained to take place at those lipid rafts that are concentrated in cholesterol. Because of the depleted cholesterol supply, there are fewer lipid rafts, and this interferes with glucose uptake. Glucose and fats are the main sources of energy for muscles, and both are compromised.
As I mentioned earlier, statins interfere with the synthesis of coenzyme Q10 (Langsjoen and Langsjoen, 2003), which is highly concentrated in the heart as well as the skeletal muscles, and, in fact, in all cells that have a high metabolic rate. It plays an essential role in the citric acid cycle in mitochondria, responsible for the supply of much of the cell’s energy needs. Carbohydrates and fats are broken down in the presence of oxygen to produce water and carbon dioxide as by-products. The energy currency produced is adenosine triphosphate (ATP), and it becomes severely depleted in the muscle cells as a consequence of the reduced supply of coenzyme Q10.
The muscle cells have a potential way out, using an alternative fuel source, which doesn’t involve the mitochondria, doesn’t require oxygen, and doesn’t require insulin. What it requires is an abundance of fructose in the blood, and fortunately (or unfortunately, depending on your point of view) the liver’s statin-induced impairment results in an abundance of serum fructose. Through an anaerobic process taking place in the cytoplasm, specialized muscle fibers skim off just a bit of the energy available from fructose, and produce lactate as a product, releasing it back into the blood stream. They have to process a huge amount of fructose to produce enough energy for their own use. Indeed, statin therapy has been shown to increase the production of lactate by skeletal muscles (Pinieux et al, 1996).
Converting one fructose molecule to lactate yields only two ATP’s, whereas processing a sugar molecule all the way to carbon dioxide and water in the mitochondria yields 38 ATP’s. In other words, you need 19 times as much substrate to obtain an equivalent amount of energy. The lactate that builds up in the blood stream is a boon to both the heart and the liver, because they can use it as a substitute fuel source, a much safer option than glucose or fructose. Lactate is actually an extremely healthy fuel, water-soluble like a sugar but not a glycating agent.
So the burden of processing excess fructose is shifted from the liver to the muscle cells, and the heart is supplied with plenty of lactate, a high-quality fuel that does not lead to destructive glycation damage. LDL levels fall, because the liver can’t keep up with fructose removal, but the supply of lactate, a fuel that can travel freely in the blood (does not have to be packaged up inside LDL particles) saves the day for the heart, which would otherwise feast off of the fats provided by the LDL particles. I think this is the crucial effect of statin therapy that leads to a reduction in heart attack risk: the heart is well supplied with a healthy alternative fuel.
This is all well and good, except that the muscle cells get wrecked in the process. Their cell walls are depleted in cholesterol because cholesterol is in such short supply, and their delicate fats are therefore vulnerable to oxidation damage. This problem is further compounded by the reduction in coenzyme Q10, a potent antioxidant. The muscle cells are energy starved, due to dysfunctional mitochondria, and they try to compensate by processing an excessive amount of both fructose and glucose anaerobically, which causes extensive glycation damage to their crucial proteins. Their membranes are leaking ions, which interferes with their ability to contract, hindering movement. They are essentially heroic sacrificial lambs, willing to die in order to safeguard the heart.
Muscle pain and weakness are widely acknowledged, even by the statin industry, as potential side effects of statin drugs. Together with a couple of MIT students, I have been conducting a study which shows just how devastating statins can be to muscles and the nerves that supply them (Liu et al, 2011). We gathered over 8400 on-line drug reviews prepared by patients on statin therapy, and compared them to an equivalent number of reviews for a broad spectrum of other drugs. The reviews for comparison were selected such that the age distribution of the reviewers was matched against that for the statin reviews. We used a measure which computes how likely it would be for the words/phrases that show up in the two sets of reviews to be distributed in the way they are observed to be distributed, if both sets came from the same probability model. For example, if a given side effect showed up a hundred times in one data set and only once in the other, this would be compelling evidence that this side effect was representative of that data set. Table 1 shows several conditions associated with muscle problems that were highly skewed towards the statin reviews.
Side Effect # Statin Reviews # Non-Statin Reviews Associated P-value Muscle Cramps 678 193 0.00005 General Weakness 687 210 0.00006 Muscle Weakness 302 45 0.00023 Difficulty Walking 419 128 0.00044 Loss of Muscle Mass 54 5 0.01323 Numbness 293 166 0.01552 Muscle Spasms 136 57 0.01849 Table 1: Counts of the number of reviews where phrases associated with various symptoms related to muscles appeared, for 8400 statin and 8400 non-statin drug reviews, along with the associated p-value, indicating the likelihood that this distribution could have occurred by chance.
I believe that the real reason why statins protect the heart from a heart attack is that muscle cells are willing to make an incredible sacrifice for the sake of the larger good. It is well acknowledged that exercise is good for the heart, although people with a heart condition have to watch out for overdoing it, walking a careful line between working out the muscles and overtaxing their weakened heart. I believe, in fact, that the reason exercise is good is exactly the same as the reason statins are good: it supplies the heart with lactate, a very healthy fuel that does not glycate cell proteins.
Membrane Cholesterol Depletion and Ion Transport
As I alluded to earlier, statin drugs interfere with the ability of muscles to contract through the depletion of membrane cholesterol. (Haines, 2001) has argued that the most important role of cholesterol in cell membranes is the inhibition of leaks of small ions, most notably sodium (Na+) and potassium (K+). These two ions are essential for movements, and indeed, cholesterol, which is absent in plants, is the key molecule that permits mobility in animals, through its strong control over ion leakage of these molecules across cell walls. By protecting the cell from ion leaks, cholesterol greatly reduces the amount of energy the cell needs to invest in keeping the ions on the right side of the membrane.
There is a widespread misconception that “lactic acidosis,” a condition that can arise when muscles are worked to exahustion, is due to lactic acid synthesis. The actual story is the exact opposite: the acid build-up is due to excess breakdown of ATP to ADP to produce energy to support muscle contraction. When the mitochondria can’t keep up with energy consumption by renewing the ATP, the production of lactate becomes absolutely necessary to prevent acidosis (Robergs et al., 2004). In the case of statin therapy, excessive leaks due to insufficient membrane cholesterol require more energy to correct, and all the while the mitochondria are producing less energy.
In in vitro studies of phospholipid membranes, it has been shown that the removal of cholesterol from the membrane leads to a nineteen fold increase in the rate of potassium leaks through the membrane (Haines, 2001). Sodium is affected to a lesser degree, but still by a factor of three. Through ATP-gated potassium and sodium channels, cells maintain a strong disequilibrium across their cell wall for these two ions, with sodium being kept out and potassium being held inside. This ion gradient is what energizes muscle movement. When the membrane is depleted in cholesterol, the cell has to burn up substantially more ATP to fight against the steady leakage of both ions. With cholesterol depletion due to statins, this is energy it doesn’t have, because the mitochondria are impaired in energy generation due to coenzyme-Q10 depletion.
Muscle contraction itself causes potassium loss, which further compounds the leak problem introduced by the statins, and the potassium loss due to contraction contributes significantly to muscle fatigue. Of course, muscles with insufficient cholesterol in their membranes lose potassium even faster. Statins make the muscles much more vulnerable to acidosis, both because their mitochondria are dysfunctional and because of an increase in ion leaks across their membranes. This is likely why athletes are more susceptible to muscle damage from statins (Meador and Huey, 2010, Sinzinger and O’Grady, 2004): their muscles are doubly challenged by both the statin drug and the exercise.
An experiment with rat soleus muscles in vitro showed that lactate added to the medium was able to almost fully recover the force lost due to potassium loss (Nielsen et al, 2001). Thus, production and release of lactate becomes essential when potassium is lost to the medium. The loss of strength in muscles supporting joints can lead to sudden uncoordinated movements, overstressing the joints and causing arthritis (Brandt et al., 2009). In fact, our studies on statin side effects revealed a very strong correlation with arthritis, as shown in the table.
While I am unaware of a study involving muscle cell ion leaks and statins, a study on red blood cells and platelets has shown that there is a substantial increase in the Na+-K+-pump activity after just a month on a modest 10 mg/dl statin dosage, with a concurrent decrease in the amount of cholesterol in the membranes of these cells (Lohn et al., 2000). This increased pump activity (necessitated by membrane leaks) would require additional ATP and thus consume extra energy.
Muscle fibers are characterized along a spectrum by the degree to which they utilize aerobic vs anaerobic metabolism. The muscle fibers that are most strongly damaged by statins are the ones that specialize in anaerobic metabolism (Westwood et al., 2005). These fibers (Type IIb) have very few mitochondria, as contrasted with the abundant supply of mitochondria in the fully aerobic Type 1A fibers. I suspect their vulnerability is due to the fact that they carry a much larger burden of generating ATP to fuel the muscle contraction and to produce an abundance of lactate, a product of anaerobic metabolism. They are tasked with both energizing not only themselves but also the defective aerobic fibers (due to mitochondrial dysfunction) and producing enough lactate to offset the acidosis developing as a consequence of widespread ATP shortages.
Long-term Statin Therapy Leads to Damage Everywhere
Statins, then, slowly erode the muscle cells over time. After several years have passed, the muscles reach a point where they can no longer keep up with essentially running a marathon day in and day out. The muscles start literally falling apart, and the debris ends up in the kidney, where it can lead to the rare disorder, rhabdomyolysis, which is often fatal. In fact, 31 of our statin reviews contained references to “rhabdomyolysis” as opposed to none in the comparison set. Kidney failure, a frequent consequence of rhabdomyolysis, showed up 26 times among the statin reviews, as opposed to only four times in the control set.
The dying muscles ultimately expose the nerves that innervate them to toxic substances, which then leads to nerve damage such as neuropathy, and, ultimately Amyotrophic Lateral Sclerosis (ALS), also known as Lou Gehrig’s disease, a very rare, debilitating, and ultimately fatal disease which is now on the rise due (I believe) to statin drugs. People diagnosed with ALS rarely live beyond five years. Seventy-seven of our statin reviews contained references to ALS, as against only 7 in the comparison set.
As ion leaks become untenable, cells will begin to replace the potassium/sodium system with a calcium/magnesium based system. These two ions are in the same rows of the periodic table as sodium/potassium, but advanced by one column, which means that they are substantially larger, and therefore it’s much harder for them to accidentally leak out. But this results in extensive calcification of artery walls, heart valves, and the heart muscle itself. Calcified heart valves can no longer function properly to prevent backflow, and diastolic heart failure results from increased left ventricular stiffness. Research has shown that statin therapy leads to increased risk to diastolic heart failure (Silver et al., 2004, Weant and Smith, 2005). Heart failure shows up 36 times in our statin drug data as against only 8 times in the comparison group.
Once the muscles can no longer keep up with lactate supply, the liver and heart will be further imperilled. They’re now worse off than they were before statins, because the lactate is no longer available, and the LDL, which would have provided fats as a fuel source, is greatly reduced. So they’re stuck processing sugar as fuel, something that is now much more perilous than it used to be, because they are depleted in membrane cholesterol. Glucose entry into muscle cells, including the heart muscle, mediated by insulin, is orchestrated to occur at lipid rafts, where cholesterol is highly concentrated. Less membrane cholesterol results in fewer lipid rafts, and this leads to impaired glucose uptake. Indeed, it has been proposed that statins increase the risk to diabetes (Goldstein and Mascitelli, 2010, Hagedorn and Arora, 2010). Our data bear out this notion, with the probability of the observed distributions of diabetes references happening by chance being only 0.006.
Side Effect # Statin Reviews # Non-Statin Reviews Associated P-value Rhabdomyolysis 31 0 0.02177 Liver Damage 326 133 0.00285 Diabetes 185 62 0.00565 ALS 71 7 0.00819 Heart Failure 36 8 0.04473 Kidney Failure 26 4 0.05145 Arthritis 245 120 0.01117 Memory Problems 545 353 0.01118 Parkinson’s Disease 53 3 0.01135 Neuropathy 133 73 0.04333 Dementia 41 13 0.05598 Table 2: Counts of the number of reviews where phrases associated with various symptoms related to major health issues appeared, besides muscle problems, for 8400 statin and 8400 non-statin drug reviews, along with the associated p-value, indicating the likelihood that this distribution could have occurred by chance.
Statins, Caveolin, and Muscular Dystrophy
Lipid rafts are crucial centers for transport of substances (both nutrients and ions) across cell membranes and as a cell signaling domain in essentially all mammalian cells. Caveolae (“little caves”) are microdomains within lipid rafts, which are enriched in a substance called caveolin (Gratton et al., 2004). Caveolin has received increasing attention of late due to the widespread role it plays in cell signaling mechanisms and the transport of materials between the cell and the environment (Smart et al., 1999).
Statins are known to interfere with caveolin production, both in endothelial cells (Feron et al., 2001) and in heart muscle cells, where they’ve been shown to reduce the density of caveolae by 30% (Calaghan, 2010). People who have a defective form of caveolin-3, the version of caveolin that is present in heart and skeletal muscle cells, develop muscular dystrophy as a consequence (Minetti et al., 1998). Mice engineered to have defective caveolin-3 that stayed in the cytoplasm instead of binding to the cell wall at lipid rafts exhibited stunted growth and paralysis of their legs (Sunada et al., 2001). Caveolin is crucial to cardiac ion channel function, which, in turn, is essential in regulating the heart beat and protecting the heart from arrhythmias and cardiac arrest (Maguy et al, 2006). In arterial smooth muscle cells, caveolin is essential to the generation of calcium sparks and waves, which, in turn, are essential for arterial contraction and expansion, to pump blood through the body (Taggart et al, 2010).
In experiments involving constricting the arterial blood supply to rats’ hearts, researchers demonstrated a 34% increase in the amount of caveolin-3 produced by the rat’s hearts, along with a 27% increase in the weight of the left ventricle, indicating ventricular hypertrophy. What this implies is that the heart needs additional caveolin to cope with blocked vessels, whereas statins interfere with the ability to produce extra caveolin (Kikuchi et al., 2005).
Statins and the Brain
While the brain is not the focus of this essay, I cannot resist mentioning the importance of cholesterol to the brain and the evidence of mental impairment available from our data sets. Statins would be expected to have a negative impact on the brain, because, while the brain makes up only 2% of the body’s weight, it houses 25% of the body’s cholesterol. Cholesterol is highly concentrated in the myelin sheath, which encloses axons which transport messages long distances (Saher et al., 2005). Cholesterol also plays a crucial role in the transmission of neurotransmitters across the synapse (Tong et al, 2009). We found highly skewed distribution of word frequencies for dementia, Parkinson’s disease, and short term memory loss, with all of these occurring much more frequently in the statin reviews than in the comparison reviews.
A recent evidence-based article (Cable, 2009) found that statin drug users had a high incidence of neurological disorders, especially neuropathy, parasthesia and neuralgia, and appeared to be at higher risk to the debilitating neurological diseases, ALS and Parkinson’s disease. The evidence was based on careful manual labeling of a set of self-reported accounts from 351 patients. A mechanism for such damage could involve interference with the ability of oligodendrocytes, specialized glial cells in the nervous system, to supply sufficient cholesterol to the myelin sheath surrounding nerve axons. Genetically-engineered mice with defective oligodendrocytes exhibit visible pathologies in the myelin sheath which manifest as muscle twitches and tremors (Saher et al, 2005). Cognitive impairment, memory loss, mental confusion, and depression were also significantly present in Cable’s patient population. Thus, his analysis of 351 adverse drug reports was largely consistent with our analysis of 8400 reports.
Cholesterol’s Benefits to Longevity
The broad spectrum of severe disabilities with increased prevalence in statin side effect reviews all point toward a general trend of increased frailty and mental decline with long-term statin therapy, things that are usually associated with old age. I would in fact best characterize statin therapy as a mechanism to allow you to grow old faster. A highly enlightening study involved a population of elderly people who were monitored over a 17 year period, beginning in 1990 (Tilvis et al., 2011). The investigators looked at an association between three different measures of cholesterol and manifestations of decline. They measured indicators associated with physical frailty and mental decline, and also looked at overall longevity. In addition to serum cholesterol, a biometric associated with the ability to synthesize cholesterol (lathosterol) and a biometric associated with the ability to absorb cholesterol through the gut (sitosterol) were measured.
Low values of all three measures of cholesterol were associated with a poorer prognosis for frailty, mental decline and early death. A reduced ability to synthesize cholesterol showed the strongest correlation with poor outcome. Individuals with high measures of all three biometrics enjoyed a 4.3 year extension in life span, compared to those for whom all measures were low. Since statins specifically interfere with the ability to synthesize cholesterol, it is logical that they would also lead to increased frailty, accelerated mental decline, and early death.
For both ALS and heart failure, survival benefit is associated with elevated cholesterol levels. A statistically significant inverse correlation was found in a study on mortality in heart failure. For 181 patients with heart disease and heart failure, half of those whose serum cholesterol was below 200 mg/dl were dead three years after diagnosis, whereas only 28% of the patients whose serum cholesterol was above 200 mg/dl had died. In another study on a group of 488 patients diagnosed with ALS, serum levels of triglycerides and fasting cholesterol were measured at the time of diagnosis (Dorstand et al., 2010). High values for both lipids were associated with improved survival, with a p-value < 0.05.
What to do Instead to Avoid Heart Disease
If statins don’t work in the long run, then what can you do to protect your heart from atherosclerosis? My personal opinion is that you need to focus on natural ways to reduce the number of small dense LDL particles, which feed the plaque, and alternative ways to supply the product that the plaque produces (more about that in a moment). Obviously, you need to cut way back on fructose intake, and this means mainly eating whole foods instead of processed foods. With less fructose, the liver won’t have to produce as many LDL particles from the supply side. From the demand side, you can reduce your body’s dependency on both glucose and fat as fuel by simply eating foods that are good sources of lactate. Sour cream and yogurt contain lots of lactate, and milk products in general contain the precursor lactose, which gut bacteria will convert to lactate, assuming you don’t have lactose intolerance. Strenuous physical exercise, such as a tread machine workout, will help to get rid of any excess fructose and glucose in the blood, with the skeletal muscles converting them to the much coveted lactate.
Finally, I have a set of perhaps surprising recommendations that are based on research I have done leading to the two papers that are currently under review (Seneff3 et al, Seneff4 et al.). My research has uncovered compelling evidence that the nutrient that is most crucially needed to protect the heart from atherosclerosis is cholesterol sulfate. The extensive literature review my colleagues and I have conducted to produce these two papers shows compellingly that the fatty deposits that build-up in the artery walls leading to the heart exist mainly for the purpose of extracting cholesterol from glycated small dense LDL particles and synthesizing cholesterol sulfate from it, providing the cholesterol sulfate directly to the heart muscle. The reason the plaque build-up occurs preferentially in the arteries leading to the heart is so that the heart muscle can be assured an adequate supply of cholesterol sulfate. In our papers, we develop the argument that the cholesterol sulfate plays an essential role in the caveolae in the lipid rafts, in mediating oxygen and glucose transport.
The skin produces cholesterol sulfate in large quantities when it is exposed to sunlight. Our theory suggests that the skin actually synthesizes sulfate from sulfide, capturing energy from sunlight in the form of the sulfate molecule, thus acting as a solar-powered battery. The sulfate is then shipped to all the cells of the body, carried on the back of the cholesterol molecule.
Evidence of the benefits of sun exposure to the heart is compelling, as evidenced by a study conducted to investigate the relationship between geography and cardiovascular disease (Grimes et al., 1996). Through population statistics, the study showed a consistent and striking inverse linear relationship between cardiovascular deaths and estimated sunlight exposure, taking into account percentage of sunny days as well as latitude and altitude effects. For instance, the cardiovascular-related death rate for men between the ages of 55 and 64 was 761 in Belfast, Ireland but only 175 in Toulouse, France.
Cholesterol sulfate is very versatile. It is water soluble so it can travel freely in the blood stream, and it enters cell membranes ten times as readily as cholesterol, so it can easily resupply cholesterol to cells. The skeletal and heart muscle cells make good use of the sulfate as well, converting it back to sulfide, and synthesizing ATP in the process, thus recovering the energy from sunlight. This decreases the burden on the mitochondria to produce energy. The oxygen released from the sulfate molecule is a safe source of oxygen for the citric oxide cycle in the mitochondria.
So, in my view, the best way to avoid heart disease is to assure an abundance of an alternative supply of cholesterol sulfate. First of all, this means eating foods that are rich in both cholesterol and sulfur. Eggs are an optimal food, as they are well supplied with both of these nutrients. But secondly, this means making sure you get plenty of sun exposure to the skin. This idea flies in the face of the advice from medical experts in the United States to avoid the sun for fear of skin cancer. I believe that the excessive use of sunscreen has contributed significantly, along with excess fructose consumption, to the current epidemic in heart disease. And the natural tan that develops upon sun exposure offers far better protection from skin cancer than the chemicals in sunscreens.
Concluding Remarks
Every individual gets at most only one chance to grow old. When you experience your body falling apart, it is easy to imagine that this is just due to the fact that you are advancing in age. I think the best way to characterize statin therapy is that it makes you grow older faster. Mobility is a great miracle that cholesterol has enabled in all animals. By suppressing cholesterol synthesis, statin drugs can destroy that mobility. No study has shown that statins improve all-cause mortality statistics. But there can be no doubt that statins will make your remaining days on earth a lot less pleasant than they would otherwise be.
To optimize the quality of your life, increase your life expectancy, and avoid heart disease, my advice is simple: spend significant time outdoors; eat healthy, cholesterol-enriched, animal-based foods like eggs, liver, and oysters; eat fermented foods like yogurt and sour cream; eat foods rich in sulfur like onions and garlic. And finally, say “no, thank-you” to your doctor when he recommends statin therapy.
References
[1] K.D. Brandt, P. Dieppe, E. Radin, “Etiopathogenesis of osteoarthritis”. Med. Clin. North Am. 93 (1): 1–24, 2009.
[2] J. Cable, “Adverse Events of Statins – An Informal Internet-based Study,” JOIMR, 7(1), 2009. [3] S. Calaghan, “Caveolae as key regulators of cardiac myocyte beta2 adrenoceptor signalling: a novel target for statins” Research Symposium on Caveolae: Essential Signalosomes for the Cardiovascular System, Proc Physiol Soc 19, SA21, University of Manchester, 2010.
[4] K.S. Collison, S.M. Saleh, R.H. Bakheet, R.K. Al-Rabiah, A.L. Inglis, N.J. Makhoul, Z.M. Maqbool, M. Zia Zaidi, M.A. Al-Johi and F.A. Al-Mohanna, “Diabetes of the Liver: The Link Between Nonalcoholic Fatty Liver Disease and HFCS-55” Obesity, 17(11), 2003-2013, Nov. 2009.
[5] J. Dorstand, P. Ku ̈hnlein, C. Hendrich, J. Kassubek, A.D. Sperfeld, and A.C. Ludolph. “Patients with elevated triglyceride and cholesterol serum levels have a prolonged survival in amyotrophic lateral sclerosis,” J Neurol. in Press:Published online Dec. 3 2010.
[6] O. Feron, C. Dessy, J.-P. Desager, andJ.-L. Balligand, “Hydroxy-Metholglutaryl-Coenzyme A Reductase Inhibition Promotes Endothelial Nitric Oxide Synthase Activation Through a Decrease in Caveolin Abundance,” Circulation 103, 113-118, 2001.
[7] M.R. Goldstein and L. Mascitelli, “Statin-induced diabetes: perhaps, its the tip of the iceberg,” QJM, Published online, Nov 30, 2010.
[8] S.S. Gottlieb, M. Khatta, and M.L. Fisher. “Coenzyme Q10 and congestive heart failure.” Ann Intern Med, 133(9):745–6, 2000.
[9] J.-P. Gratton, P. Bernatchez, and W.C. Sessa, “Caveolae and Caveolins in the Cardiovascular System,” Circulation Research, 94:1408-1417, June 11, 2004.
[10] D.S. Grimes, E. Hindle and T. Dyer, “Sunlight, Cholesterol and Coronary Heart Disease,” Q. J. Med 89, 579-589, 1996; http://www.ncbi.nlm.nih.gov/pubmed/8935479
[11] J. Hagedorn and R. Arora, “Association of Statins and Diabetes Mellitus,” American Journal of Therapeutics, 17(2):e52, 2010.
[12] T.H. Haines, “Do Sterols Reduce Proton and Sodium Leaks through Lipid Bilayers?” Progress in Lipid Research, 40, 299-324., 2001; http://www.ncbi.nlm.nih.gov/pubmed/11412894
[13] T. Kikuchi, N. Oka, A. Koga, H. Miyazaki, H. Ohmura, and T. Imaizumi, “Behavior of Caveolae and Caveolin-3 During the Development of Myocyte Hypertrophy,” J Cardiovasc Pharmacol. 45:3, 204-210, March 2005.
[14] P.H. Langsjoen and A.M. Langsjoen, “The clinical use of HMG CoA-reductase inhibitors and the associated depletion of coenzyme Q10. A review of animal and human publications.” Biofactors, 18(1):101–111, 2003.
[15] J. Liu, A. Li and S. Seneff, “Automatic Drug Side Effect Discovery from Online Patient-Submitted Reviews: Focus on Statin Drugs.” Submitted to First International Conference on Advances in Information Mining and Management (IMMM) Jul 17-22, 2011, Bournemouth, UK.
[16] M. Löhn, M. Fürstenau, V. Sagach, M. Elger, W. Schulze, F.C. Luft, H. Haller, and M. Gollasch, “Ignition of Calcium Sparks in Arterial and Cardiac Muscle Through Caveolae,” Circ. Res. 2000;87;1034-1039
[17] A. Maguy, T.E. Hebert, and S. Nattel, “Involvement of Lipid rafts and Caveolae in cardiac ion channel function,” Cardiovascular Research, 69, 798-807, 2006.
[18] B.M. Meador and K.A. Huey, “Statin-Associated Myopathy and its Exacerbation with Exercise,” Muscle and Nerve, 469-79, Oct. 2010.
[19] C. Minetti, F. Sotgia, C. Bruno, et al., “Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy,” Nat. Genet., 18, 365-368, 1998.
[20] O.B. Nielsen, F. de Paoli, and K. Overgaard, “Protective effects of lactic acid on force production in rat skeletal muscles.” J. Phhsiology 536(1), 161-166, 2001.
[21] P.S. Phillips, R.H. Haas, S. Bannykh, S. Hathaway, N.L. Gray, B.J. Kimura, G. D. Vladutiu, and J.D.F. England. “Statin-associated myopathy with normal creatine kinase levels,” Ann Intern Med, October 1, 2002;137:581–5.
[22] G. de Pinieux, P. Chariot, M. Ammi-Said, F. Louarn, J.L. LeJonc, A. Astier, B. Jacotot, and R. Gherardi, “Lipid-lowering drugs and mitochondrial function: effects of HMG-CoA reducase inhibitors on serum ubiquinone and blood lactate/pyruvate ratios.” Br. J. Clin. Pharmacol. 42: 333-337, 1996.
[23] R.A. Robergs, F. Ghiasvand, and D. Parker, “Biochemistry of exercise-induced metabolic acidosis.” Am J Physiol Regul Integr Comp Physiol 287: R502–R516, 2004.
[24] G. Saher, B. Brügger, C. Lappe-Siefke, et al. “High cholesterol level is essential for myelin membrane growth.” Nat Neurosci 8:468-75, 2005.
[25] S. Seneff, G. Wainwright, and L. Mascitelli, “Is the Metabolic Syndrome Caused by a High Fructose, and Relatively Low Fat, Low Cholesterol Diet?” Archives of Medical Science, 7(1), 8-20, 2011; DOI: 10.5114/aoms.2011.20598
[26] S. Seneff, G. Wainwright, and L. Mascitelli, “Nutrition and Alzheimer’s Disease: the Detrimental Role of a High Carbohydrate Diet,” In Press, European Journal of Internal Medicine, 2011.
[27] S. Seneff, G. Wainwright and B. Hammarskjold, “Cholesterol Sulfate Supports Glucose and Oxygen Transport into Erythrocytes and Myocytes: a Novel Evidence Based Theory,” submitted to Hypotheses in the Life Sciences.
[28] S. Seneff, G. Wainwright and B. Hammarskjold, “Atherosclerosis may Play a Pivotal Role in Protecting the Myocardium in a Vulnerable Situation,” submitted to Hypotheses in the Life Sciences.
[29] H. Sinzinger and J. O’Grady, “Professional athletes suffering from familial hypercholesterolaemia rarely tolerate statin treatment because of muscle problems.” Br J Clin Pharmacol 57,525-528, 2004.
[30] E.J. Smart, G.A. Graf, M.A. McNiven, W.C. Sessa, J.A. Engelman, P.E. Scherer, T. Okamoto, and M.P. Lisanti, “Caveolins, Liquid-Ordered Domains, and Signal Transduction,” Molecular and Cellular Biology, 19, 7289–7304, Nov. 1999.
[31] A.J. Shyam Kumar, S.K. Wong, and G. Andrew, “Statin-induced muscular symptoms : A report of 3 cases.” Acta Orthop. Belg. 74, 569-572, 2008.
[32] M.A. Silver, P.H. Langsjoen, S. Szabo, H. Patil, and A. Zelinger, “Effect of atorvastatin on left ventricular diastolic function and ability of coenzyme Q10 to reverse that dysfunction.” The American Journal of Cardiology, 94(10):1306–1310, 2004.
[33] Y. Sunada, H. Ohi, A. Hase, H. Ohi, T. Hosono, S. Arata, S. Higuchi, K. Matsumura, and T. Shimizu, “Transgenic mice expressing mutant caveolin-3 show severe myopathy associated with increased nNOS activity,” Human Molecular Genetics 10(3) 173-178, 2001. http://hmg.oxfordjournals.org/content/10/3/173.abstract
[34] M. J. Taggart, “The complexity of caveolae: a critical appraisal of their role in vascular function,” Research Symposium on Caveolae: Essential Signalosomes for the Cardiovascular System, Proc Physiol Soc 19, SA21, University of Manchester, 2010.
[35] P. Thavendiranathan, A.Bagai, M.A. Brookhart, and N.K. Choudhry, “Primary prevention of cardiovascular diseases with statin therapy: a meta-analysis of randomized controlled trials,” Arch Intern Med. 166(21), 2307-13., Nov 27, 2006.
[36] R.S. Tilvis, J.N. Valvanne, T.E. Strandberg and T.A. Miettinen “Prognostic significance of serum cholesterol, lathosterol, and sitosterol in old age; a 17-year population study,” Annals of Medicine, Early Online, 1–10, 2011.
[37] J. Tong, P.P. Borbat, J.H. Freed, and Y. Shin, “A scissors mechanism for stimulation of SNARE-mediated lipid mixing by cholesterol.” Proc Natl Acad Sci U S A 2009;106:5141-6.
[38] L. Vila, A. Rebollo, G.S. AÄ‘alsteisson, M. Alegret, M. Merlos, N. Roglans, and J.C. Laguna, “Reduction of liver fructokinase expression and improved hepatic inflammation and metabolism in liquid fructose-fed rats after atorvastatin treatment,” Toxicology and Applied Pharmacology 251, 32–40, 2011.
[39] Walley T., Folino-Gallo P., Stephens P et al, “Trends in prescribing and utilisation of statins and other lipid lowering drugs across Europe 1997-2003,” Br J Clin Pharmacol 60, 543-551, 2005.
[40] K.A. Weant and K.M. Smith, “The Role of Coenzyme Q10 in Heart Failure,” Ann Pharmacother, 39(9), 1522-6, Sep. 2005.
[41] F. R. Westwood, A. Bigley, K. Randall, A.M. Marsden, and R.C. Scott, “Statin-induced muscle necrosis in the rat: distribution, development, and fibre selectivity,” Toxicologic Pathology, 33:246-257, 2005.
Stephanie Seneff can be contacted by email at seneff@csail.mit.edu
Related articles









STATIN NATION
View another excerpt from the documentary film Statin Nation.
For more information, please visit www.statinnation.net
Related videos
Related article
- Bad Pharma And The Statin Wars (cardiobrief.org)
Featured Video

Press freedom threatened as US government seizes Max Blumenthal’s devices
or go to
Aletho News Archives – Video-Images
From the Archives
Roosevelt Conspired to Start World War II in Europe
By John Wear – Inconvenient History – January 26, 2019
Establishment historians claim that U.S. President Franklin D. Roosevelt never wanted war and made every reasonable effort to prevent war. This article will show that contrary to what establishment historians claim, Franklin Roosevelt and his administration wanted war and made every effort to instigate World War II in Europe. … continue
Blog Roll
 Aletho News
Aletho News- Pakistan thought it won the peace. Now, it could be dragged into war.
- Trump’s bombing campaign in Iran just sinks U.S. into a deeper quagmire
- US Threats Against Iran’s Nuclear Facilities Violate International Law – Iranian Foreign Ministry
- Ukrainian human rights commissioner warns of growing public anger at forced conscription
- The Zumwalt-class destroyer is an ever-growing boondoggle
- Andy Burn ’Em, Britain’s new PM, wants to end homelessness by fueling militarism and war
- China hawks are losing the public opinion battle
- Press Freedom Threatened as U.S. Government Seizes Max Blumenthal’s Devices
- Henrik Svensmark fired by his university
- Yemeni naval blockade forces six vessels to turn back
- If Americans Knew
- Series of Reports Ignored by Media Show Jeffrey Epstein’s Extensive Work With Israeli Intelligence
- Babies in Gaza Die Preventable Deaths Due to Medical Shortages Imposed by Israel
- AG Complaint Filed: Israel Interfering in US Elections Thru Brad Parscale
- Gaza family of six, killed by Israeli missiles as they slept – Daily Update
- Bethlehem: Every morning the same question: “Why are we still here?”
- Israel’s War Kills More U.S. Troops
- Lethal Precision Without Accountability: Israeli Government “Quadcopter” Use in Gaza
- ‘Sip your coffee while watching the sunrise’: How Israel is colonizing the West Bank by selling Palestinian land to Jewish Americans
- Pro-Israel GOP donors fueling Rep. Stevens’ Dem primary in Michigan
- Israel is building a Genocide Wall in Gaza – Daily Update
- No Tricks Zone
- Global Temperature Trend Has Cooled Over The Past 6500 Years, Scientists Have Found
- Wind Energy Means Going Back To The Middle Ages, Says German Professor Horst-Joachim Lüdecke
- New Study: A 40-Fold Increase In Earth’s Main Greenhouse Gas Contributes To Cooling The Ocean
- New Study Highlights The ‘Dominant Role’ Of Aerosol/Cloud Interactions In Shaping Climate
- Munich’s First-Ever Green Party Mayor Declares First Ever City Water Use Restrictions… Fines Up to 50,000 €!
- Experimental Lab Research: The Climate Sensitivity To A 400-Fold Increase In CO2 Is 0.1°C
- Fatal Snobbery: In France, It’s Better To Die From A Heatwave Than To Do As Americans
- New Study: NASA’s Models Wildly Underestimate The Capacity Of Clouds To Alter Solar Radiation
- Polar Freezeover: Western Arctic Early July Sea Ice Exceeds 1980s Average
- Doing The Opposite: Studies Show Gigantic Wind Farms Significantly Warm The Night
